Untangling the life sciences
(PhysOrg.com) -- Last month, Dr. Michael Stadler and his Computational Biology group at the Friedrich Miescher Institute for Biomedical Research became a member laboratory of the Swiss Institute of Bioinformatics. This is in well-deserved recognition of the widely appreciated expertise in computational biology residing in Michael Stadler and his FMI bioinformatics team.
There must be a genetic pattern that triggers breast cells to transform and become cancerous. There are likely to be expression differences between identical twins, despite their matching genes. There should be a switch that changes the expression patterns in cells during differentiation. However, finding the meaningful patterns, differences and switches in the tangle of information we can obtain from cells, is a major challenge. For over forty years, approaching this tangle by sequencing gene products has been a promising avenue. But with ever faster and more powerful sequencing methods, the data supply from genes, RNAs, and genome-maps of epigenetic marks, have reached gargantuan dimensions. Obtaining and handling millions of sequences is no longer an issue, but disentangling them in a meaningful way requires a more rare and essential art - the art of the computational biologist.
"We are helping biologists to understand better how the different layers of epigenetic, transcriptional and post-transcriptional regulation interact and contribute to the control of gene expression", comments Michael Stadler, Head of the Computational Biology Group at the Friedrich Miescher Institute for Biomedical Research (FMI). His team not only contributes in an innovative and creative manner to the research efforts of many groups at the FMI, but they constantly develop new analytical methods and software tools that allow the interpretation of large datasets generally. Indeed, the team conducts their own bioinformatics research, much of which has been exploited for data processing pipelines in Novartis and elsewhere. In recognition of the team's widely acknowledged expertise, Michael Stadler was named a full member of the Swiss Institute of Bioinformatics (SIB) at the SIB Foundation Council Meeting earlier this month. The SIB, which exists since the late 1990's, unites under its virtual roof the leading groups in bioinformatics from all Swiss universities. SIB "plays a federating role, linking the bioinformatics community and promoting and coordinating research and education throughout Switzerland".
Give us large datasets
One of the strengths of Stadler's team is the analysis of data from next generation sequencing and from genome wide datasets. "With the recent technological advances it has become possible to quantify components of the gene expression system genome-wide, but also for individual cells," said Stadler. Dirk Schübeler, Group Leader at the FMI, has profited from this expertise to study differences in the methylation patterns on chromatin and their influence on the expression pattern in cells. "Our results challenge the simple division of chromatin into active euchromatin and inactive heterochromatin. The findings reveal a more complex pattern of genome accessibility linked to histone modifications. We have been able to draw such a conclusion thanks to the sound and strong bioinformatics evidence, provided by Michael Stadler and his team."
Resolving patterns
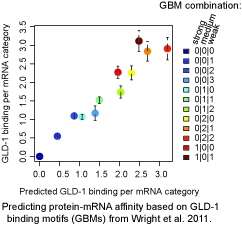
A computational biology approach has also led to the identification of RNA motifs recognized by a conserved RNA-binding protein, GLD-1, and to explaining how their combinations determine the affinity of GLD-1 for its mRNA targets. This study has been published earlier this year in the EMBO Journal. Rafal Ciosk, FMI group leader and responsible for this study explains, "In this study we used a ribonomics approach to understand how a key regulator of germ cell development, GLD-1, interacts with the transcriptome". GLD-1 binds and represses the translation of selected mRNAs and thereby controls many aspects of germ line development, for example maintenance of the totipotent state of germ cells. The "ribonomic" computational analysis was able to predict precisely how GLD-1 interacts with ~1000 target RNAs. Remarkably, this study demonstrates that transcriptome-wide identification of mRNA targets of an RNA-binding protein, combined with quantitative computational analysis, can generate highly predictive models of post-transcriptional regulatory networks.
Apart from these excursions into the tangles, it is the everyday bioinformatics services that the team provides to PhD students and postdocs alike that makes it extremely valuable for the FMI. "Sequence comparisons, homologue searches, data mining of the easier kind are part of the things we do on a day-to-day basis and that benefit many projects at the FMI," summarizes Stadler. The recognition of their support appears in nearly every talk given by FMI members, underscoring the importance bioinformatics plays in cutting-edge biomedical research.
More information: Wright JE, et al. (2011) A quantitative RNA code for mRNA target selection by the germline fate determinant GLD-1. EMBO J 30, 533 - 545 (02 February 2011) doi:10.1038/emboj.2010.334
Provided by Friedrich Miescher Institute for Biomedical Research