Researchers develop new strategy to uncover structural variations of human genomes
The study on single-nucleotide resolution structural variations (SVs) of an Asian and African genome was published online in Nature Biotechnology. This study was performed by BGI (previously known as the Beijing Genomics Institute), the largest genomics organization in the world, and demonstrates that whole genome de novo assembly could serve as a new solution for developing a more comprehensive SV map of individuals.
With the rapid development of genomics, more and more experts focus upon the studies of human genome variations by identification and annotation of SNPs in the context of structure, function, and disease. However, recent studies have shown that there are a large number of SVs that have been discovered in the human genome putatively having equal or greater functional impacts than SNPs.
Although many methods have been used to characterize SVs in previous studies, each may have some disadvantages due to technological limitations and the complexity of SVs, making it necessary to find a high accuracy detection method to identify and characterize SVs of human genomes.
"The research focusing on SVs is a real challenge," said Yingrui Li, Director of Science and Technology Department at BGI and the co-lead author of the study, "The study was confronted with many difficulties at the start, such as alignment accuracy, rearranged structure (non-linear), breakpoint recovery, and background noises."
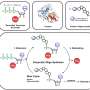
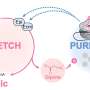
"As a solution," he explained, 'researchers discovered a novel pipeline for detecting SVs in Whole Genome Assembly with a lower cost and faster speed." Based on large-scale genome assembly data from next-generation sequencing technologies, small and intermediate size homozygous SVs (1- 50kbp) can be detected, including insertions, deletions, inversions, and complex rearrangements with precise breakpoints and genotypes previously difficult to define by other approaches.
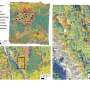
Through this new method, researchers identified 277,243 SVs, ranging from 1bp to 23kbp in assembled regions of both genomes. Meanwhile, the researchers performed validation using computational and experimental methods and the results indicated a high accuracy of detection. They also carried out characterization of genome-wide patterns of these SVs on different genomic features and studied their potential biological impacts. Profiling using 106 individuals of the 1000 Genomes Project indicates that the extent of diversity in SVs between individuals exceeds that of SNPs. These findings demonstrate whole genome de novo assembly could serve as a new solution to a more comprehensive SV map.
"Here we provide a new method, at a relatively low cost and high speed, to establish in greater detail the presence and patterns of SVs in different genomes, and the results have a high accuracy and a wider range of length spectrum coverage in comparison with previous methods," said Honglong Wu, bioinformatician at BGI and one senior author of the study.
Furthermore, researchers reported, SVs are more individual-specific than SNPs, which may play a significant role underlying the phenotypic differences between individuals. "This study makes us understand we need to consider all kinds of genetic variations and potential differences in their impacts on disease and various other phenotypes in medical genomics studies in the future." added Yingrui Li.
Professor Jun Wang, Executive Director of BGI, said, "With further progresses in de novo assembling by new technologies, assembly-based approaches will be of greater importance and potentially an ultimate solution to SV determination. The study of SVs is likely to attract even more attention in the future."
This study also reveals that de novo assembly can develop more complete personal genomes than resequencing based mapping. Researchers recommend using de novo sequencing technology to decode many more human genomes in the future.
Provided by Beijing Genomics Institute