Computer program takes on protein puzzle
(PhysOrg.com) -- All proteins self-assemble in a fraction of the blink of an eye, but it can take a long time to mimic the process. And there has been no guarantee of success, even with the most powerful computers - until now.
Rice University researchers have come up with a computer program to accurately simulate protein folding dramatically faster than previous methods. It will allow scientists to peer deeper into the roots of diseases caused by proteins that fold incorrectly.
Authors Cheng Zhang and Jianpeng Ma describe their simulation of three short proteins with the new technique in the cover story of the current Journal of Chemical Physics. Ma is a professor in the Department of Bioengineering at Rice University and the Department of Biochemistry and Molecular Biology at Baylor College of Medicine. Zhang is an applied physics graduate student at Rice.
"Protein folding is regarded as one of the biggest unsolved problems in biophysics," Ma said. "This is a technically challenging task, and many groups around the world have been competing for years to make the process faster and more accurate."
Understanding the intricacies of protein folding is a crucial step in deciphering the genetic code that serves as the operating system of all living things.
Correctly folded proteins perform many roles: as enzymes vital to metabolism; structural elements in bone, muscle and cell scaffolding; mechanisms in cell signaling and immune response and much more. But protein misfolding is a critical factor in many diseases, including Alzheimer's, cystic fibrosis, emphysema and various cancers.
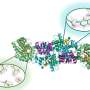
Proteins start as amino acid molecules floating in a cell. Following DNA blueprints, the molecules are strung together like beads on a necklace, called a polypeptide. Every polypeptide of a given sequence will fold precisely the same way into the shape, called the native state, that determines its function.
Like a river finding the shortest route to the sea, proteins always find their way to their native states in an instant. How that happens is one of life's great mysteries. "The question is how nature finds this final folded state so quickly," Ma said.
Zhang and Ma reached unprecedented accuracy and speed in simulating the folding of three relatively short but well-understood proteins -- trpzip2, trp-cage and the villin headpiece -- in the presence of water molecules, which Ma described as the best way to simulate physiological conditions.
Though the proteins assemble themselves in nature almost instantly, the Rice team's algorithm took weeks to run the simulation. Still, that was far faster than others have achieved. "And for trpzip and villin, nobody has reached this level of accuracy in the native state under similar simulation conditions -- that is, in the presence of water, which is the most stringent condition," Ma said.
The researchers employed two novel strategies, continuously variable temperature and single-copy simulation.
"In the process of simulation, called sampling, the computer has to search through many, many possible structures of the protein chain to find the lowest-energy solution," Ma said. "A polypeptide chain en route to its native state encounters many energy barriers, much like when one navigates through a rugged mountain landscape.
"Speeding up the process of crossing those barriers is the key to finding the true global minimum (energy state)," he said. "In our simulation, temperature is a variable that goes continuously up and down. When the temperature is higher, proteins can overcome energy barriers faster. It's equivalent to speeding up the motion of atoms."
Ma said the previous state of the art was to run multiple copies of a simulation in parallel on many computers -- an intensive and expensive approach. "The single-copy approach uses only one simulation, essentially, to find the native state of the protein. This is a major plus, because anyone with reasonable computing power can run this method."
Even so, it takes computational muscle to simulate a biological task that the body's cells accomplish as a matter of course. Zhang and Ma found it in Rice's supercomputer cluster, the Shared University Grid at Rice, aka SUG@R. "We can't overstate the significance of state-of-the-art computing facilities, as well as excellent service from Rice's Research Computing Support Group," Ma said. His group is continuing its work on Rice's newest supercomputer cluster, BlueBioU, for longer polypeptide sequences.
"These supercomputer resources will continue to make Rice one of the leading institutions in the field of protein folding and computational biophysics," Ma said.
More information: Read the abstract here: jcp.aip.org/jcpsa6/v132/i24/p244101_s1
Provided by Rice University