Genome of Irish potato famine pathogen decoded

(PhysOrg.com) -- A large international research team has decoded the genome of the notorious organism that triggered the Irish potato famine in the mid-19th century and now threatens this season's tomato and potato crops across much of the US.
Published in the September 9 online issue of the journal Nature, the study reveals that the organism boasts an unusually large genome size — more than twice that of closely related species — and an extraordinary genome structure, which together appear to enable the rapid evolution of genes, particularly those involved in plant infection. These data expose an unusual mechanism that enables the pathogen to outsmart its plant hosts and may help researchers unlock new ways to control it.
"This pathogen has an exquisite ability to adapt and change, and that's what makes it so dangerous," said senior author Chad Nusbaum, co-director of the Genome Sequencing and Analysis Program at the Broad Institute of MIT and Harvard. "We now have a comprehensive view of its genome, revealing the unusual properties that drive its remarkable adaptability. Hopefully, this knowledge can foster novel approaches to diagnose and respond to outbreaks."
"Our findings suggest a 'two-speed' genome, meaning that different parts of the genome are evolving at different rates," said co-lead author Sophien Kamoun, head of the Sainsbury Laboratory in Norwich, UK. "Future sequencing of additional strains and close relatives of this pathogen will help test this hypothesis and could transform our understanding of how it adapts to immune plants."
The potato famine that gripped Europe, particularly Ireland, in the mid 1800's was the work of an insidious organism known as Phytophthora infestans. Long considered a fungus, it is now known to be a member of the oomycetes or "water molds," which are more closely related to the malaria parasite than to fungi. P. infestans thrives in cool, wet weather, and can infect potatoes, tomatoes and other related plants, causing a "late blight" disease that can decimate entire fields in just a few days.
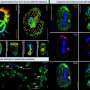
, P. sojae (yellow) and P. ramorum (red). The multicoloured rim shows the attacking proteins (effectors), comprised of a number of domains. Some of these have remained unchanged during evolution (blue triangles and green rhombi), whilst others are highly variable (other forms and colours). The diversity in effector proteins and the dynamics of the Phytophthora genome contribute to P. infestans’ ability to circumvent late blight resistance genes in potato. (Source: Haas et al. Nature, 9 September 2009)")
Not only swift in its destruction, the pathogen is also remarkable in its ability to change. For example, it can quickly adapt to new plant hosts, attacking even genetically resistant potatoes that have been painstakingly bred to fend off P. infestans infection. How the pathogen can adapt so rapidly to these immune potatoes has long puzzled scientists.
To understand the genetic basis for the pathogen's adaptive success, the researchers, led by scientists at the Broad Institute and the Sainsbury Laboratory, decoded the P. infestans genome. They produced a high-quality genome sequence and compared it to the genomes of two relatives: P. sojae, which infects soybeans, and P. ramorum, which prefers oak and other trees and causes a condition known as sudden oak death.
One of the most striking findings to emerge from these comparisons is the expanded nature of the P. infestans genetic blueprint: It is two and a half to four times the size of its relatives' genomes.
But perhaps even more surprising than the genome's large size is the source of its added bulk. Nusbaum and his colleagues determined that the additional genomic real estate does not reflect more genes per se, but instead stems from a massive expansion in the amount of repetitive (once considered to be "junk") DNA. In fact, this type of DNA accounts for about 75% of the entire P. infestans genome.
"Such a large amount of repetitive DNA is pretty surprising, since there is a metabolic cost to maintain it," said Nusbaum. "As a genome biologist, I have to wonder how the organism benefits from having it."
The researchers gained some key insights into the potential advantages of carrying this glut of repetitive DNA by probing its genomic structure. They made three critical observations:
- The P. infestans genome comprises alternating repeat-rich (and gene-poor) regions and gene-dense regions;
- These gene-dense regions are shared among other Phytophthora species, preserved over millions of years of evolution, whereas the repeat-rich regions are undergoing relatively rapid changes;
- The repeat-rich regions contain fewer genes compared to other genomic regions, yet those genes they do contain are enriched for those that play crucial roles in plant infection.
Taken together, these findings suggest an unusual genomic strategy to support the rapid evolution of critical genes, known as "effector" genes. Effector genes can disrupt plants' normal physiology, enabling the pathogen to establish a foothold. However, some can also trigger plants' immune responses, making them prime targets for combating P. infestans infection.
"We think this could be a tactic that enables P. infestans to rapidly adapt to host plants," said co-lead author Brian Haas, manager of genome annotation, outreach, bioinformatics, and analysis at the Broad Institute. "In contrast to the well-conserved regions where most genes are found, the repeat-rich regions change rapidly over time, acting as a kind of incubator to enable the rapid birth and death of genes that are key to plant infection. As a result, these critical genes may be gained and lost so rapidly that the hosts simply can't keep up."
Importantly, the new P. infestans genome sequence enabled the researchers to identify many previously unknown effector genes, particularly those that belong to two key groups, known as RXLR genes and CRN genes. The research team identified more than 500 RXLR genes and nearly 200 CRN genes, significantly more than are found in the pathogen's relatives.
These findings not only expand the catalog of known P. infestans genes, they also highlight a critical subset of genes undergoing rapid turnover. Further studies of these genes will foster a deeper understanding of plant infection and help identify potential targets for fighting back.
More information: Haas et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature DOI:10.1038/nature08358
Source: Broad Institute of MIT and Harvard