January 4, 2021 report
Stopping SARS-CoV-2 in its tracks by blocking its main protease

Inasmuch as therapeutic options against coronavirus have been focused mainly on blocking the interaction between its spike protein and the ACE2 receptor on host cells, SARS-CoV-2 has several additional critical proteins that could potentially be targeted with drugs that have already been approved for use against other viruses. One of these viral proteins is the main protease (Mpro) that is needed to separate newly minted polypeptides into their functional component parts.
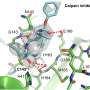
Scientists from the Department of Biophysics at the All India Institute of Medical Sciences have recently put several promising inhibitors of Mpro through the paces to see what sticks. Their findings, appearing in the American Chemical Society's journal ACS Omega, suggest that an HIV drug known as cobicistat is looking pretty good. But what exactly constitutes a "good" drug here, and how do researchers even find them in the first place?
A nice starting point is to have some idea of what your target looks like. In this case, the 3-D structure of Mpro had recently become available to serve as the basis for what is known in the business as rational structure-based drug design. To get things going, the researchers virtually screened the approved drugs compound library over at the Drug Bank pharmaceutical knowledge base to find possible inhibitors of Mpro. Typically, this involves performing molecular docking studies to shortlist the best candidates. One of the most common metrics used in this endeavor is to look for molecules with a high glide docking score and glide energy.
A GlideScore is computed using software like Glide from Schrodinger. In a nutshell, it ranks so-called "poses" of different ligands by simulating binding free energy; the more negative the value, the tighter the binding. This empirical scoring function includes terms for force field contributions (electrostatic, van der Waals) and also terms rewarding or penalizing other interactions known to influence ligand binding. Typically, up to 300 atoms and 50 rotatable bonds can be simulated, which is plenty for small molecule drugs or even peptide ligands up to about 11 residues.
The authors also used a more advanced method of calculated binding energy of ligand-protein complexes known as molecular mechanics-Poisson Boltzmann surface area (MM-PBSA). This method combines energetic calculations based on molecular mechanics with free energy calculations based on implicit solvent models. Stated another way, it estimates the binding free energy of a ligand-protein complex as the difference between the free energy of the complex and the free energies of the unbound components, including both entropic and enthalpic terms.
The next step in the drug discovery process is usually to do more detailed molecular dynamic simulations to further pigeonhole candidate molecules and reduce the number that need to be experimentally tested. MD simulation refines the interactions of docked complexes by provisioning for protein flexibility and detailed solvent effects. The outputs of MD simulations are represented as graphs of RMSD (root-mean-square deviation) of atomic position fluctuations of the protein backbone, or radius of gyration, and also number of hydrogen bonds, as functions of the run length, given in nanoseconds.
When all is said and done, what the authors are really after is the actual true-to-life molecular interaction kinetics of their chosen molecules. Only real measurements can provide this, and as we discussed a few days ago, there are several new kinds of instruments that can pull this off. Perhaps the most handy method to divine molecular interaction kinetics is through SPR (surface plasmon resonance). SPR delivers sensorgrams from which the association rate constants (kon) and dissociation rate constants (koff) for the binding of a potential drug inhibitor to a target like Mpro can be determined. One manufacturer of SPR instruments, Nicoya, has an excellent blog post that describes in more detail how these assays work.
The authors were able to determine the equilibrium dissociation constants, KD (M), for potential inhibitors like cobicistat, since it is easily derived from the kinetic data the relation: KD = koff/kon. The only thing left to do after identifying cobicistat as the winner of the drug candidate primary was to verify that it inhibited Mpro in an enzyme activity assay. For drugs that might potentially block interaction between the SARS-CoV-2 spike protein and its ACE2 receptor, the inhibitor might be applied to the SPR assay as a 'disrupter' of the binding of ligand to target. Kinetic data would not be readily obtained in this kind of situation.
As the target of Mpro has not been ported to an SPR-friendly application, the authors measured the inhibition of Mpro activity using a universal protease assay. They were able to derive an IC50 value for cobicistat of ∼6.7 μM, which was more favorable than that obtained for the other potential drugs cangrelor and denufosol (0.9 mM, and 1.3 mM, respectively). The IC50 indicates how much drug is needed to inhibit activity by 50%.
The cleavage site where Mpro acts was also found to be different from the cleavage site where the many extant human proteases act. This is fortunate, because any drug that blocks our own proteases would undoubtedly have significant side effects. The other SARS-CoV-2 protease, PLpro, recognizes an important molecule known as ubiquitin and any attempts to inhibit this protease might be expected to wreak havoc on our own critical deubiquitinase systems.
In the spirit of defeating all things COVID, some further interesting developments have taken place within the larger sphere of vaccine readiness, namely, in decoding how, and therefore whether, these new vaccines might be expected to work. Bert Hubert has led the public charge to try to figure out how Pfizer has optimized their mRNA vaccine for translation in our cells. In particular, he has created a programming challenge to find the supposed algorithm with which the codon optimization of the ~4000 character-long vaccine was performed.
This assumes, of course, that some actual algorithm, rather than just hand-crafted individual codon tinkering was done, in which case, the shortest algorithm wouldn't be very short at all. In fact, it would be pretty much the same algorithm that created the entire universe that evolved into Pfizer. The latest updates, which includes user-submitted algorithms that predict the actual codons with over 90% accuracy from the incipient hacker-turned-biologist community can be found over on Bert's Pfizer vaccine reverse engineering site.
On a final note, it seems that other competing mRNA vaccines like those from Moderna or CureVac are likely to be very similar to the Pfiver vaccine. For example, a fellow named Pavol Rusnak just extracted the CureVac sequence to a text file and posted it here. While these codons encode much the same amino-acids as BNT162b2, 33% of the codons are different. Although it can be tough to keep track of everything going on with the larger SARS-CoV-2 ecosystem, one incredible technical wellspring should escape the attention of no serious observer and this is the social media account of Ersa Flavinkins, AKA @flavinkins. Be sure to tune in for all the latest facts, theories, speculations, and of course, conspiracies.
More information: Akshita Gupta et al. Structure-Based Virtual Screening and Biochemical Validation to Discover a Potential Inhibitor of the SARS-CoV-2 Main Protease, ACS Omega (2020). DOI: 10.1021/acsomega.0c04808
Journal information: ACS Omega
© 2021 Science X Network