Biomedical engineers develop computational model to better understand genomes
Biomedical engineers have developed a computational model that will help biological researchers clearly identify the significance of variations between different genomes — the complex sequences of DNA and RNA at the foundation of all living organisms. The findings will be published March 31 in the open-access journal PLoS Computational Biology.
The international team of researchers, from the University of Virginia, USA, Wageningen University, The Netherlands, and Helmholtz Center for Infection Research, Germany, demonstrated their approach by focusing on the pathogen Pseudomonas aeruginosa—a bacterium that causes about 10 percent of hospital-acquired infections. The bacterium is especially problematic for burn victims and those with cystic fibrosis or those whose immune systems are compromised, and this new approach is an important first step toward improving their treatment.
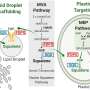
In recent years, researchers have been mapping the genomes of multiple organisms and they can now measure the activity of specific genes across the entire genome at the same time and under multiple environments. The emerging field of systems biology integrates this information into computational models. While these models can be used to predict which genes are critical for various cell functions—such as how the cells will respond to medicine or how fast they will grow in different environments—there are still limitations to the science.
"Unfortunately, as these models get built, some of the differences between the models of two cells or two bacteria can be an artifact of the model-building process itself," said Jason Papin, one of the authors and an assistant professor of biomedical engineering at University of Virginia. "Our paper presents an approach for reconciling two models so that you can have confidence that the differences are actually present in the living systems. With the reconciled models, you can then start to ask very specific questions like which genes that are unique to each bacterium are essential for some basic processes."
To illustrate the efficacy of their approach, the researchers compared the pathogen Pseudomonas aeruginosa—one of the principal antibiotic resistant bacteria—with the non-pathogen Pseudomonas putida. The reconciled models clarified how the explicit differences between the genomes of the bacteria mapped to differences in functions of the cells.
More information: Oberhardt MA, Puchałka J, Martins dos Santos VAP, Papin JA (2011) Reconciliation of Genome-Scale Metabolic Reconstructions for Comparative Systems Analysis. PLoS Comput Biol 7(3): e1001116. doi:10.1371/journal.pcbi.1001116
Provided by Public Library of Science