Simulations unravel outer membrane transport mechanism
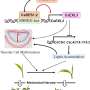
 and the luminal domain (in green and dark blue) of the outer membrane transporter. The yellow sphere represents the point on the TonB protein at which the force was applied. To view the results of the simulation, please go to: http://www.life.uiuc.edu/emad/TonB-BtuB/btub-2.5Ans.mpg . Credit: Image courtesy of Emad Tajkhorshid")
Using X-ray data and advanced computer simulation and visualization software, researchers at the University of Illinois have painstakingly modeled a critical part of a mechanism by which bacteria take up large molecules. Their findings provide a rare window on the complex interplay of proteins involved in the active transport of materials across cell membranes.
Their study, which includes a collaborator from the University of Virginia, appears online in the Biophysical Journal and was described in the May 25 edition of Science.
Transporting large molecules, such as vitamin B12, citric acid or other vital nutrients across the outer membranes of Gram-negative bacteria is not a simple task. The cells must be selective in what substances they take up, and the outer membrane contains no energy-generating machinery to power the job of hauling large molecules inside.
The new study examined an outer membrane transport system that depends on an energy-generating inner membrane protein, TonB. This TonB-dependent transporter (TBDT) contains a beta-barrel domain: a series of parallel sheets that form a tunnel through which large molecules can pass. Another region of the protein, the luminal domain, clogs this barrel until the cell is ready to allow large molecules to pass through.
Crystallographic studies had shown that TonB binds to one end of the luminal domain. Researchers had hypothesized that TonB somehow draws the luminal domain out of the barrel or changes its conformation to make way for the large molecules.
Previous studies had been inconclusive, however. Molecular biologists have difficulty studying systems that involve complex interactions between proteins, particularly when one domain moves into and out of a structure like a beta-barrel, said biochemistry professor Emad Tajkhorshid, principal investigator on the study. “It’s very difficult to assess this experimentally because they have to look at the accessibility of certain (amino acids) before and after activation,” Tajkhorshid said. “And sometimes the labeling agent that they use diffuses into the beta-barrel and labels the inside: You don’t know whether what you have labeled is happening inside or outside.”
To address these limitations, Tajkhorshid and graduate student James Gumbart used two software programs developed in the U. of I.’s National Institutes of Health Resource for Macromolecular Modeling and Bioinformatics. The programs, NAMD and VMD, respectively, simulate and visualize complex molecular interactions. By entering detailed data about the position and characteristics of every atom in the system, the researchers ran simulations of various scenarios to test which hypotheses were most feasible. Their work relied on detailed crystallographic studies of the molecules provided by University of Virginia researcher Michael C. Wiener.
“The good thing about simulations is that you can monitor the position of every atom,” Tajkhorshid said.
The task was enormous, however.
“The fundamental motions of atoms that guide large conformational changes happen on a very short time scale: femtoseconds,” Tajkhorshid said. Each step relies on the completion of a previous step, so the simulations take an extensive amount of time and considerable computational effort.
The researchers addressed two key questions: First, could the bond between TonB and the luminal domain withstand the force needed to pull the luminal domain downward, away from the barrel" Second, how does the luminal domain respond to force in order to expose a permeation pathway through the barrel"
In the first simulation the researchers applied a force to TonB and showed that the multiple hydrogen bonds between TonB and the luminal domain were strong enough to remain intact while TonB pulled the end of the luminal domain away from the barrel. The simulation also showed the luminal domain gradually unfolding, changing its conformation in ways that would open up enough space for a molecule of vitamin B12 to pass through the barrel. (Click on the link below the image of the system to watch the results of the simulation.)
A second simulation exerted a force near the center of gravity of the luminal domain, pulling it out of the barrel in one piece. This required an enormous input of energy, however.
“The force that we applied was about 4,000 picoNewtons,” Tajkhorshid said. “This is an order of magnitude higher than that required to induce the unfolding. This clearly indicates that the unplugging mechanism is very unlikely.”
Coupled with increasingly sophisticated crystallographic studies and basic biological data, simulations help researchers study the interactions of multiple proteins in complex systems, such as the TonB-dependent transporter, Gumbart said.
“As we get more computational power, we’re looking less at single proteins and more and more at the interaction of proteins,” he said. “And this is going in the direction of what’s called systems biology. This is essential because none of these proteins actually works alone. Each works in concert with many other things.”
Tajkhorshid is also a professor of pharmacology in the College of Medicine and an affiliate of the Beckman Institute and the Center for Biophysics and Computational Biology in the College of Liberal Arts and Sciences.
Source: University of Illinois at Urbana-Champaign