Texas A&M researchers develop tool to study complex clusters of genes
Texas A&M University researchers have developed a computational tool that will help scientists more accurately study complex units of clustered genes, called operons, in bacteria.
The tool, which allows scientists to analyze many bacterial genomes at once, is more accurate than previous methods because it starts from experimentally validated data instead of from statistical predictions, they say. The researchers hope their tool will lead to a better understanding of the complex genetic mechanisms involved in a cell's functioning.
"It's a very complicated mechanism inside a cell that makes the whole thing work, and operons are one of the important components in this process," said Sing-Hoi Sze, Texas A&M computer science, biochemistry and biophysics researcher. "We want to understand how these genetic mechanisms work because DNA codes proteins, and proteins are what make up everything in your body. In order to understand the genetic processes in more complex organisms, we have to start with the simpler organisms like bacteria."
Sze and his colleague, computer science researcher Qingwu Yang, detail their computational tool and its implications in their paper published in the journal Genome Research.
An operon is a unit of genes that are clustered together and have similar functions, Sze said. Genes are controlled by a mechanism called a promoter, which turns the genes on or off. In higher organisms, like humans, there is usually a specific promoter that controls each gene separately, Sze explained. However, a bacteria's genome has to be compact, so there are a lot of genes clustered close together that are controlled by the same promoter, and this set of genes is called an operon.
Different species of bacteria have similar genes, but their genes may not have the same layout or clustering pattern, so their operons may function differently, Sze said. Scientists want to understand how the operons in each species of bacteria are different, how the genes in the operons are organized, and how the operons function.
Because of time and resource constraints, however, researchers cannot directly study in detail the genomes of all of the thousands of species of bacteria and can only carry out experiments on some of them, Sze explained. So researchers need a computational tool to help them predict where similar clusters of genes are in different species of bacteria so they can better focus their experiments, he said.
For each level of complexity in organisms, there is a model organism that scientists center their experiments on, and for bacteria, the model organism is E. coli. Because E. coli is a model organism, scientists have studied it in great detail and have a large amount of experimentally validated data on its genome and the operons that function, Sze said.
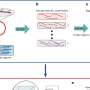
Sze and Yang's computational tool starts with a known and experimentally validated E. coli operon and then searches each of hundreds of separate species of bacteria for genes that are related to those in the E. coli operon. Once the tool has located related genes in a bacterium, it then checks to see if there is a strong clustering of the genes. It does this by using a statistical procedure that computes which genes are statistically very close to each other when you compare it to a random situation of genes, Sze explained.
"Imagine putting genes in a random order," Sze said. "When placed randomly, two specific genes are likely to be located at a far distance from each other. But if there is clustering, then the genes will be much closer to each other than they would be by chance."
So by using Sze and Yang's tool, scientists can easily locate sub-blocks of genes in different bacteria that are arranged in a similar fashion as one of the operons in E. coli. "If biologists are interested in a particular E. coli operon, they can use our tool to find where the operon is in the different bacteria," Sze said. "They can then analyze the differences in the operon in the different bacteria and see if there are any interesting relationships."
Sze said his and Yang's tool is an improvement on previous methods because it is a new way to analyze many bacterial genomes at the same time. It is also more accurate than previous methods in which scientists start by simply using a statistical method to predict the location of an operon in a bacterial genome. Sze and Yang, however, show that it is more accurate to start from a known and experimentally validated E. coli operon to find similar operons in other bacteria.
"Eventually, we want to try to improve our tool to make it better and more accurate," Sze said. "Although our tool can analyze a lot of bacteria at the same time, it compares each bacterium to E. coli separately. So the ultimate goal would be to develop a tool that will analyze them all together."
Source: Texas A&M University