Researchers restore missing protein in rare genetic brain disorder
UCSF researchers have successfully used protease inhibitors to restore to normal levels a key protein involved in early brain development. Reduced levels of that protein have been shown to cause the rare brain disorder lissencephaly, which is characterized by brain malformations, seizures, severe mental retardation and very early death in human infants.
The findings offer a proof-of-principle, at least in mice, that the genetic equivalent to human lissencephaly, also known as "smooth brain" disease, can be treated during pregnancy and effectively reversed to produce more normal offspring. Findings are reported in the September issue of "Nature Medicine" and found online at nature.com.
While the progress is still in animal models, the work is significant in being the first successful attempt to use protease inhibitors to reverse a severe brain defect that is known to be caused by limited quantities of one key gene, the researchers say.
The hope is that this approach also could be used to treat other defects in utero, or even those manifesting after birth, when caused by a partial deficiency in one gene, according to Anthony Wynshaw-Boris, MD, PhD, who is chief of the UCSF Division of Genetics in the Department of Pediatrics, and a member of the UCSF Institute for Human Genetics.
"Researchers have not considered it possible to treat such a pervasive, early developmental brain disorder as lissencephaly," said Wynshaw-Boris, who collaborated on the paper with Shinji Hirotsune, MD, PhD, in the Osaka City University Graduate School of Medicine. "Not only were we able to show a clear cellular effect from using these protease inhibitors, but also were able to treat the disorder in utero."
The work is the culmination of 15 years of collaborative research in the Wynshaw-Boris and Hirotsune labs into the cause and mechanisms of lissencephaly, which is caused by a deletion or loss of one copy of the LIS1 gene and affects an estimated one in 50,000-100,000 infants.
In 1998, the team published a paper on work that Hirotsune did in the Wynshaw-Boris laboratory, in which he produced a mouse with the same mutation that displayed defective brain development. They have continued to collaborate on understanding the mechanism of action of LIS1 since Hirotsune set up his independent laboratory in Japan.
The current research found, using these mice, that the protein calpain degrades the LIS1 protein to less than half its normal levels near the surface of the cells. The team then used a specific small-molecule protease inhibitor of calpain in these mice. At a cellular level, the protease inhibitors enabled LIS1 protein to be expressed at near-normal levels.
The team then gave daily injections of a calpain inhibitor to pregnant mice whose fetuses had the mouse-model of this defect. The resulting offspring had more normal brains and showed no sign of mental retardation.
"This study is really a proof-of-principle not only for treating complex developmental brain disorders, but also for any disorder with reduced protein levels where proteases normally play some role in breaking down that protein," Hirotsune said. "This will be much more difficult to apply to humans, because of the safety issues involved, but it could lead to new therapies that might be effective for a wide range of developmental disorders."
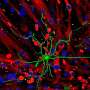
Scientists have known that loss of one of the two copies of the human form of the gene, known as LIS1, prevents immature nerve cells from migrating from deep in the brain up to the surface of the emerging cerebral cortex.
As a result, these immature cells stall at mid-point in their migration, causing the brain to develop a smooth surface, devoid of the convoluted nerve tissue that enables humans to think and function. The resulting disease, lissencephaly, varies in severity, but always leads to retardation, seizures and early childhood death.
Source: University of California - San Francisco