New potential therapeutic target discovered for genetic disorder -- Barth syndrome
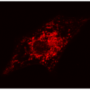
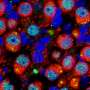
Researchers at NYU Langone Medical Center may have discovered a new targeted intervention for Barth Syndrome (BTHS). BTHS, a sometimes fatal disease, is a serious genetic disorder occurring predominantly in males that leads to infection or heart failure in childhood. The new study entitled, "Role of calcium-independent phospholipase A2 in the pathogenesis of Barth syndrome", was recently published in the Proceedings of the National Academy of Sciences, shows the benefits of targeted intervention with an iPLA2-VIA inhibitor that prevents a major symptom of the disease- cardiolipin deficiency.
"Our research has established a causal role of cardiolipin deficiency in the pathogenesis of Barth syndrome and identified an important enzyme in cardiolipin degradation called iPLA2-VIA as a potential target for therapeutic intervention of the disease," said Mindong Ren, Ph.D., lead investigator of the study and assistant professor of cell biology at NYU Langone Medical Center.
BTHS syndrome is an X-linked genetic cardioskeletal muscle disease resulting in muscle weakness and fatigue in patients. The debilitating disorder is caused by a mutation in the genetic coding of tafazzin, an enzyme of the cardiolipin pathway. Cardiolipin is an essential lipid in the inner membrane of mitochondria responsible for normal cell structure and energy production. BTHS patients exhibit defects in cardiolipin metabolism which help fight infections. The various symptoms of BTHS, in addition to cardiolipin deficiency, include cardiomyopathy (weakness in heart muscle), neutropenia (a reduction in neutrophils or white blood cells that fight bacterial infections), muscle weakness & fatigue (caused by cellular deficiency), growth delay, and increase of organic acids in urine.
In a previous study, NYU researchers documented the characteristics of a tafazzin-deficiency in a Drosophila (fruit fly) model of the disease, showing low and abnormal cardiolipin concentration, abnormal mitochondria, and poor motor function. In this new study researchers documented that tafazzin or cardiolipin deficiency in Drosophila disrupts the final stage of spermatogenesis causing male sterility. Using this fly model, the study showed that this trait of cardiolipin deficiency can be genetically suppressed by inactivating calcium-independent phospholipase A2, which prevents the degradation of cardiolipin. This method keeps cardiolipin levels normal. Researchers were also able to show that treatment of BTHS patients lymphoblasts within a tissue culture with the iPLA2-VIA inhibitor BEL partially restored the tissue cultures cardiolipin homeostasis.
"Taken together, our two findings establish a causal role of cardiolipin deficiency in the pathogenesis of Barth syndrome and identify iPLA2-VIA as a very important enzyme," said Michael Schlame, M.D., associate professor of anesthesiology and cell biology, NYU Langone Medical Center. "This is good news for patients since this enzyme is now a potential target for therapeutic intervention."
According to researchers, although this has not been tested in humans, the successful restoration of these mutated cells with BEL shows promise for continued BTHS research, patients and their families. There are no treatments for Barth syndrome at this time.
More information: www.pnas.org/content/106/7/233 … 0f-a418-ef589e10e510
Source: NYU Langone Medical Center / New York University School of Medicine