Unexpected protein interaction suggests new ALS drug target
Discovery of an unexpected protein-protein interaction has led University of Iowa scientists and colleagues to identify a drug that slows the progression of Amyotrophic Lateral Sclerosis (ALS) in mice and nearly doubles the animals' lifespan. The study is published Jan. 24 online in the Journal of Clinical Investigation.
The UI findings may lead to a treatment for some forms of ALS, and the research also reveals a biological mechanism that might represent a new drug target for ALS and other neurological diseases.
ALS, also known as Lou Gehrig's disease, is a fatal, progressive neurodegenerative disease that affects the motor nerve cells of the brain and spinal cord. Degeneration of motor neurons impairs muscle control and movement and eventually leads to paralysis and death. "Sporadic" ALS, which can affect anyone, is the most common form of disease accounting for 90 to 95 percent of all case in the United States. About 5 to 10 percent of ALS cases are inherited.
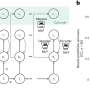
While studying the basic biology of cell signaling, scientists led by John Engelhardt, Ph.D., professor and head of anatomy and cell biology in the UI Roy J. and Lucille A. Carver College of Medicine, made the unexpected discovery that superoxide dismutase-1 (SOD1), a protein that is mutated in inherited forms of ALS, interacts with Rac1, a protein that regulates the production of reactive oxygen species (ROS) by the Nox2 protein complex.
ROS are short-lived, highly reactive molecules that are essential for normal cell functions including signaling. However, excess ROS can cause damaging oxidative stress, and abnormal ROS production has been implicated as a cause of ALS and other neurodegenerative diseases.
The unexpected interaction between a protein that is mutated in familial ALS and the cell machinery that produces ROS, which are implicated in ALS progression, prompted the scientists to investigate further.
Recently, they found that deletion of the Nox2 protein almost doubles the lifespan of mice with an inherited form of ALS, strengthening the notion that Nox2-generated ROS play a role in ALS progression. In their latest study, the researchers have shown that the drug called apocynin, which blocks Nox2, similarly slows progression and increases lifespan of ALS-affected mice.
Although the apocynin results are quite dramatic in mice, and the drug does not appear to be toxic to the animals, Engelhardt cautions that rigorous safety and efficacy testing in pre-clinical and clinical trials will be required to determine if the drug may also be useful in humans.
"We are very excited about helping to move these findings toward clinical trials," added Henry Paulson, M.D., Ph.D., neurologist and study author. "There is a great need for effective treatments in this devastating, fatal disorder."
However, both Engelhardt and Paulson, who was a UI professor of neurology during the study and now is a professor of neurology at the University of Michigan, warn that it is not known if the drug would help in forms of ALS not caused by SOD1 mutations; the so-called “sporadic forms” of ALS. This question could not be experimentally addressed since there are no mouse models for sporadic forms ALS.
Despite the potential clinical implications of this mouse study, Engelhardt is equally excited about the underlying discovery that prompted the team to test Nox2 inhibitors in ALS-affected mice.
"The discovery of the basic mechanistic interaction between SOD1 and Rac1 may be the most important discovery of my career, and it was made essentially by accident," said Engelhardt, who also is the Roy J. Carver Chair in Molecular Medicine. "It is exciting not only from the standpoint of ALS, but also because of its implications for understanding basic cell biology. Rac1 is implicated in many cellular processes including cellular migration, proliferation and differentiation, and is an important component of inflammatory disease processes."
Using a series of experiments in cells and in mice, the researchers explored the interaction between SOD1 and Rac1.
"When SOD1 binds to Rac1, the Nox2 protein produces ROS," Engelhardt explained. "However, as soon as there is an excess of ROS (hydrogen peroxide), SOD1 separates from Rac1 and the Nox2 complex stops producing ROS. In essence, SOD-1 acts like a thermostat, which senses ROS and tells the Nox2 complex when to stop producing ROS.
"In contrast, the mutant version of SOD1, which causes ALS, is not as sensitive to ROS and doesn't disengage from Rac1. With the thermostat broken, mutant SOD-1 keeps the ROS-producing furnace burning in the cell," Engelhardt said.
The results suggest that the dysfunctional interaction between mutant SOD1 and Rac1 is responsible, at least in part, for the over-production of ROS in familial ALS and the injury this causes.
Although the exact mechanism of neuronal cell death in ALS is not clear, both oxidative stress (caused by excess ROS) and overactive inflammation, which is fueled by ROS signaling, appear to play a role. Oxidative stress and inflammation also are implicated in other neurodegenerative diseases.
"Redox-activated pathways, and specifically dysregulation of these pathways, may be a component of many types of neurodegenerative disease," Engelhardt said. "Our discovery of the Rac1-SOD1 interaction may uncover new drug targets for other neurodegenerative diseases which have redox-stress components."
Source: University of Iowa