Killing drug-resistant melanoma requires combination therapy
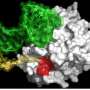
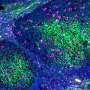
 and dead cells (blue) indicate success of a combination of MEK and PI3Ki inhibitors in killing resistant melanoma. Credit: James Hayden/The Wistar Institute")
This past summer saw a revolution in melanoma therapy. Patients whose melanoma lesions contain a mutation in the BRAF gene were successfully treated with a BRAF-specific inhibitor, PLX4032. Reports of the drug trial described shrinking tumors and improved health. Yet seven months after therapy began the tumors returned and resumed growing. Now, scientists at The Wistar Institute explain why: the tumor learns to signal around the blocked gene by adjusting its molecular wiring. They also show how to overcome resistance by simultaneously targeting multiple signaling pathways.
The researchers see this as further evidence that some cancers must be treated with multiple targeted drugs at the outset of treatment. Their findings are published in the December 14 issue of the journal Cancer Cell.
"The evidence suggests that targeting mutant BRAF can kill cancer cells, but it is not enough by itself to finish off melanoma," said Meenhard Herlyn, D.V.M., D.Sc., director of The Wistar Institute Melanoma Research Center and leader of Wistar's Molecular and Cellular Oncogenesis program. "The good news is that drugs are being developed to work in combination with BRAF inhibitors, which our data clearly shows is our best option if we intend to beat advanced melanoma."
Melanoma is the deadliest, most aggressive form of skin cancer. While surgical treatment of early melanoma leads to 90 percent cure rates, advanced melanoma is notoriously resistant to chemotherapy and has a tendency to metastasize, or spread, throughout the body. According to the World Health Organization, cases of the disease continue to rise, which has helped spur research into therapies such as BRAF inhibitors.
To study how melanoma responds to BRAF inhibitors, the Herlyn lab took melanoma cells with the BRAF mutation and tested them against a variety of anti-mutant BRAF drugs. When exposed to the drugs, the cells died off dramatically only to grow back again. In fact, cells that became resistant to one type of BRAF drug became resistant to all of them, which suggests that the cells were biochemically "rewired" in such a way that they no longer needed BRAF to form tumors.
"Cells are complex machines that work, essentially, through chains of biochemical reactions that we refer to as signaling pathways," said Jessie Villanueva, Ph.D., senior author on the study and staff scientist in the Herlyn laboratory.
"Knocking out mutant BRAF shuts a major pathway down, but if some cells can use an alternate pathway, then they can survive."
To find out which alternate pathways the drug-resistant cells use, Villanueva and her colleagues looked for signs of increased activation among proteins along the pathways BRAF uses, as well as other pathways.
Their hunt turned up two paths that worked together to aid survival. First, they found that resistant cells used a protein similar to BRAF to carry the signal down the chain. Second, they found these cells received an additional boost from the IGF-1 receptor, a protein that sits on the surface of cells and sends signals that prevent cells from being killed. The resistant cells re-route the signal around BRAF by switching to an alternate protein (CRAF or ARAF), which promotes tumor cell growth, while IGF-1R signaling promotes survival of the resistant cells.
Fortunately, there are a number compounds in clinical development that could block signals along both these pathways. So-called MEK inhibitors target a protein along the same pathway as BRAF, and IGF-1 receptor inhibitors (and inhibitors of P13K, a protein that can be activated by the IGF-1 receptor pathway) block the cancer-enabling survival signal. To test these drug combinations in the BRAF-inhibitor resistant cells, the Herlyn laboratory used a tool they developed to simulate the real-world environment of human cells: 3-D melanoma tumor spheroids. Their 3-D tissue cultures allow melanoma cells to grow in all directions, much like a new melanoma tumor would grow after metastasis. As predicted, a combination of these two inhibitors killed BRAF-resistant melanoma cells in the Wistar 3-D model.
Moreover, the Herlyn laboratory confirmed in tissue samples from patients in the PLX4032 trial—taken both before treatment and after they developed resistance—that an increased expression of the IGF-1 receptor is associated with resistance to BRAF inhibitors. None of the laboratory-generated cell lines or the post-relapse patient's tumor samples analyzed had new mutations in the BRAF, NRAS, or c-Kit genes.
Additionally, the researchers noted an association between the loss of a tumor suppressor called PTEN, and resistance to BRAF inhibitors in melanoma cell lines. The scientists found that the relapsed tumor of one patient included in the study lost the PTEN gene, even though it was present before treatment. These findings suggest that loss of PTEN could be an additional way that melanoma cells gain resistance to BRAF inhibitors. The Wistar group continues to investigate these and other mechanisms of resistance, as they expect that several will likely arise given the heterogeneous nature of melanoma.
"Tumors are efficient engines of evolution—they are going to find a way around most treatments, so we want to kill all the malignant cells from the very beginning," said Villanueva. "By targeting both pathways simultaneously you hit these cells with two punches from which they cannot recover."
"If you do this at the outset of treatment, we reason, it will prevent melanoma survival and hopefully improve patient outcomes," Villanueva added.