Screen yields drugs that could help treat fatal brain disorder
(PhysOrg.com) -- Using novel screens to sort through libraries of drugs already approved for use in human patients, a team of Wisconsin researchers has identified several compounds that could be used to treat a rare and deadly neurological disorder.
The new study, published online in the current (July 2010) issue of the journal Human Molecular Genetics by a team led by University of Wisconsin-Madison neuroscientist Albee Messing, identifies a set of FDA-approved drugs that seem to tamp down the overproduction of a brain protein that is the hallmark of Alexander disease, a fatal neurodegenerative disorder that affects mostly infants and children.
The new study, carried out during the past five years, provides a glimmer of hope for alleviating a congenital brain disease with existing drugs. Because the disease is so rare, known to have occurred in fewer than 300 people worldwide, the prospects of developing dedicated drugs to treat the condition are almost nonexistent, making such pharmaceutical prospecting the quickest path to effective treatments.
"What we are hoping for is something we can use to manage the disease and improve survival," says Messing, a Waisman Center researcher and a professor of comparative biosciences in the UW-Madison School of Veterinary Medicine. "There really are compounds out there that might be effective in manipulating gene expression in the brain."
First described in 1949, Alexander disease is caused by mutations in a gene called GFAP that encodes the production of a protein by the same name (glial fibrillary acidic protein). Although GFAP occurs in the normal central nervous system, its exact function is not well understood. It is known to be associated with astrocytes, star-shaped cells in the brain and spinal cord that perform a variety of key functions.
It was Messing's lab that originally identified GFAP as the causative gene for Alexander disease in 2001. But the Wisconsin neuroscientist now thinks that the disease is more than just the production of a mutant protein.
"We think the over-expression of GFAP is a big part of the picture," says Messing, whose lab is one of the few in the world that concentrates on the rare brain disease. "The protein aggregates affect a variety of pathways and we think there is a toxic threshold, so anything you can do to diminish production of the protein would be beneficial."
The most common form of the disease occurs in infants and is characterized by seizures, stunted motor skills and delays in cognitive development. Progressive enlargement of the head is often the most visible manifestation of the disease.
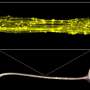
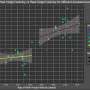
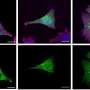
The assays used by Messing and his team were developed through the use of mouse models. High throughput screening was used to test the effects of 2880 existing drugs on purified cultures of mouse astrocytes. Ten drugs were shown to be effective, reducing the expression of the gene in a range of 37 percent to 86 percent. One of the drugs, clomipramine, commonly used to treat obsessive-compulsive disorder in humans, was tested in mice for three weeks and caused a nearly 50 percent reduction of GFAP levels in the brain.
Messing cautions that the research was still in its early stages and that further testing in animals would be necessary before a clinical trial can be contemplated. Reliable blood or spinal cord fluid markers to assess any drug's effectiveness in humans also need to be developed, and Messing's laboratory recently initiated an international collaborative study to identify such a marker. Control samples are already in hand, and enrollment of patients is actively under way.
"Without a biomarker, we're not in a position to conduct a clinical trial," says Messing. "But I'm cautiously optimistic. I think we're going to have more than one compound we can test."