Pancreatic cancer study reveals mechanism initiating disease, in mice
UCSF scientists have discovered how a mutated gene known as Kras is able to hijack mouse cells damaged by acute pancreatitis, putting them on the path to becoming pancreatic cancer cells.
The finding, they say, suggests one way in which the mutated gene -- found in nearly all cases of the most common and lethal form of pancreatic cancer -- exacts its toll in humans. It also strengthens evidence that chronic pancreatitis, which occurs when digestive enzymes become activated too soon, causing abdominal pain, indigestion and other symptoms, may be a risk factor in the development of pancreatic cancer, rather than a side effect.
The finding, says senior author Matthias Hebrok, PhD, the Hurlbut-Johnson Distinguished Professor in Diabetes Research and interim director of the UCSF Diabetes Center, could help scientists in their search for diagnostic markers in the blood of pancreatic cancer, as well as targets for therapy.
While evidence from patient tissue and mouse studies has indicated that pancreatic ductal adenocarcinoma (PDA) evolves from cells known as ductal cells -- which line the ducts through which digestive enzymes are transported from the pancreas to the small intestine -- recent studies by other labs have shown that PDA can emerge from so-called acinar cells, which produce digestive enzymes.
Subsequently, scientists in other labs have confirmed that mutated Kras can initiate the development of precursors to PDA in acinar cells. However, the findings have been puzzling, because expression of mutated Kras does not guarantee acinar reprogramming.
Given this inconsistency, scientists have hypothesized that other events in the cellular environment must occur for mutated Kras to assert itself, and recent studies have indicated that acute and chronic pancreatitis may be one such factor. When mutant Kras is active in the acinar cells of mice with chemically-induced acute pancreatitis, the development of precursors of PDA progression is accelerated.
In acute and chronic pancreatitis, acinar cells of the pancreas are persistently inflamed and injured. Under normal conditions in mouse studies, these damaged cells rapidly regenerate, in a process that involves temporarily reactivating embryonic developmental molecular signals that allow the cells to regenerate. However, in acinar cells in which mutant Kras has been activated, the embryonic developmental signals remain active, keeping the cells in a so-called "de-differentiated" state, causing them to develop into cells that are precursors to PDA.
In the current study, reported in a recent issue of The Journal of Clinical Investigation (vol. 120, issue 2, 2010), the team set out to investigate how mutant Kras co-opts the damaged acinar cells.
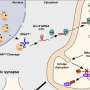
The investigation, led by John P. Morris IV, a graduate student in the Hebrok lab, examined two sets of mice. One set of normal, or "control" mice, was chemically induced to have acute pancreatitis. Another set of mice was genetically engineered to carry a copy of the mutated, or overactive, Kras gene, and then induced to develop acute pancreatitis.
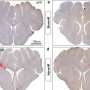
The scientists tracked the behavior, or "fate," of the fluorescently labeled acinar cells in both sets of mice. In the normal mice, the acinar cells transiently induced embryonic signaling and regenerated as expected. However, in the genetically engineered mice, the mutant Kras gene's protein, KRAS, blocked acinar cell regeneration and promoted persistently de-differentiated PDA precursor cells.
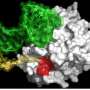
Probing for the molecular mechanism underlying this switch, the team discovered that the mutated KRAS blocked the action of a protein know as beta catenin. They also showed that beta catenin was required for efficient acinar regeneration. Most notably, they showed that when beta-catenin's signaling pathways were forcibly maintained in the context of the KRAS mutation, acinar cells did not become PDA precursors.
"These results indicate that beta-catenin is essential for acinar regeneration," says Hebrok, a member of the UCSF Helen Diller Comprehensive Cancer Center. "They also demonstrate that beta-catenin signaling must be blocked in order for mutant Kras to exploit the plasticity of cell fate that arises during pancreatic damage and to force these de-differentiated cells to become PDA precursors."
In the absence of injury, he says, "a mutation in Kras in an acinar cell likely would not cause the cell to undergo changes leading toward cancer, or it might cause these changes at a very slow rate. But if there is a mutation in Kras in a cell that has become de-differentiated in response to acute or chronic pancreatitis, this progression toward cancer would occur relatively quickly."
The finding, he says, illuminates the fine line between regeneration and carcinogenesis in the body's cells. "Cellular plasticity is necessary to allow damaged cells to activate their embryonic programs and temporarily activate embryonic signaling pathways to regenerate themselves," says Hebrok. "But cells also need to be able to turn off these signaling pathways."
"Our study underlines the likelihood that not only must mutations be acquired in a specific sequence in order to develop PDA, but levels of developmental signaling pathways must be tightly regulated to alter normal cellular plasticity and drive neoplastic cell fates."
The finding also reveals a new role of beta catenin in cancer. While the beta catenin and KRAS proteins appear to oppose one another during tumor initiation in PDA, they work together in several types of established tumors, including later stages of pancreatic cancer.