Study provides first clear idea of how rare bone disease progresses
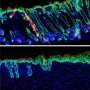
 structures at the expense of head (dorsal) structures. Credit: Mary Mullins, PhD; Shawn Little, University of Pennsylvania School of Medicine")
An international team of scientists, led by researchers at the University of Pennsylvania School of Medicine, is taking the first step in developing a treatment for a rare genetic disorder called fibrodysplasia ossificans progressiva (FOP), in which the body's skeletal muscles and soft connective tissue turns to bone, immobilizing patients over a lifetime with a second skeleton.
Reporting in the November issue of the Journal of Clinical Investigation senior authors Eileen Shore, PhD, Professor of Genetics and Orthopedics, and Mary Mullins, PhD, Professor of Cell and Developmental Biology, with scientists in Japan and Germany, demonstrated that the mutation that causes FOP mistakenly activates a cascade of biochemical events in soft tissues that kicks off the process of bone development. The linchpin of the cellular signaling gone awry is a receptor for a bone morphogenetic protein, or BMP.
The present study provides the first clear glimpse of how FOP might develop at a cellular level in the human body. Shore and co-author Frederick Kaplan, MD, the Nassau Professor of Orthopedic Molecular Medicine, and their research team, discovered the gene for FOP in 2006.
"If you think of BMP proteins as the hand that turns on a water faucet, the faucet, or receptor, should stay off if you never turn the handle," Shore says. "What our experiments show is that in FOP patients the faucet is leaky, even when it is not actively turned on." BMP receptors are protein switches that help determine the fate of stem cells in which they are expressed.
"The mutation is mildly activating, and so it may take time or the right tissue environment to allow the signal to tip the balance to induce bone formation, explains Shore. "This is a very important finding, because it can help explain why the disease progresses as it does."
The finding that the FOP mutation changes the BMP receptor such that it is effectively on most of the time gives Shore and colleagues a target to shoot for in potentially controlling the disease.
Biology Run Amok
FOP is basically a case of biology run amok. During the process of normal bone formation, a temporary cartilage structure is laid down, and then is eventually replaced by bone. In the case of FOP, that normal process of bone formation occurs inappropriately in soft tissue, sometimes in response to injury, and sometimes spontaneously, typically beginning by age 5 or so. FOP occurs in about one in 2 million individuals.
The FOP mutation is a single replacement for a DNA building block in the gene for a receptor protein called ACVR1. In 2006, Kaplan and Shore's team discovered that in the DNA of every patient with FOP they examined, the same mutation occurred: one building block in the protein-coding region of the ACVR1 gene is replaced by another, resulting in conversion of a single arginine amino acid in the sequence of the ACVR1 protein to histidine. The question the current study addresses is, what is the consequence of that change?
In experiments by Qi Shen, a postdoctoral fellow in the Shore-Kaplan lab and Shawn Little, a PhD student in the Mullins lab, the team found, using both cultured cells and zebrafish, that the specific mutation modifies ACVR1 in such a way that it acts as if it has been signaled by BMP, even when it hasn't. The experiments further show that the mutant ACVR1 receptor alters the usual binding of an ACVR1 partner protein, FKBP1A, which normally keeps the ACVR1 receptor off in the absence of BMP. The result is activation of a cell-signaling cascade that culminates in changes in gene expression, and ultimately, in the formation of new bone.
"FKBP1A is like the safety pin in a hand grenade," says Kaplan. "The FOP mutation damages the hand grenade in a very specific way that the safety pin does not work. When triggered by injury, the result is explosive new bone formation."
Enter the Zebrafish
Mullins' participation in the study was serendipitous, says Shore. Mullins studies BMP signaling in zebrafish, and in these animals BMP plays many roles, including establishing an organism's basic body plan. Mullins' long time interest was a particular gene critical to this process, called Alk8. As it turns out, Alk8 is the zebrafish equivalent of human ACVR1.
Importantly, Mullins had already established a zebrafish genetic line that fails to express Alk8. When the team inserted the gene for human ACVR1 into those fish, their normal body plan was restored. But, when they used the FOP mutation instead, the effect was one of overcompensation
"The FOP form of ACVR1 causes too much BMP expression and we get a hyper-ventralized embryo, too much cell development in the tail region of the fish," Shore explains. "So this confirmed our cell culture studies showing the mutant ACVR1 an activating mutation."
Colleagues at the Max Planck Institute for Molecular Genetics in Berlin, Germany, conducted additional experiments demonstrating that the FOP form of ACVR1 can also enhance cartilage cell differentiation. In the presence of the mutation, mild activation of cartilage development was observed to occur without activation by BMP like a leaky faucet, but could be additionally stimulated by BMP, the fully turned-on faucet.
People with FOP have a mostly normal skeleton and no evidence of extra-skeletal bone at birth; after birth it can be several years before the disease develops, forming extra-skeletal bone either spontaneously or as a result of trauma. The bone formation then progresses in a series of periodic episodes. The current study suggests this periodic progression may occur because the FOP mutation does not turn the ACVR1 faucet on all the way.
"These studies are a good beginning at getting a grasp on what the mutation is and how it is affecting BMP signaling in the cells," says Shore. "But there's a lot more to be understood."
Source: University of Pennsylvania School of Medicine (news : web)