Mitochondria could be a target for therapeutic strategy for Alzheimer's disease patients
A study in the Sept. 21 on-line edition of Nature Medicine describes the function and interaction of a critical molecule involved in cell death in Alzheimer's disease patients. These new findings reveal that blocking this molecule, called Cyclophilin D (CypD), and development of surrounding mitochondrial targets may be viable therapeutic strategies for the prevention and treatment of Alzheimer's disease, according to Shi Du Yan, Ph.D., professor of clinical pathology in the Department's of Pathology and Surgery and in the Taub Institute for Research on Alzheimer's Disease and the Aging Brain at Columbia University Medical Center, who led the multi-center research.
This paper strengthens the concept that mitochondrial permeability pores may be central in mitochondrial and neuronal malfunction relevant to Alzheimer disease. Dr. Yan and her colleagues offer new insights into the mechanism underlying amyloid beta (Aβ)-mediated mitochondrial stress through an interaction with CypD, which is linked to synaptic plasticity and learning/memory. Importantly, these findings may help explain the mechanism of action of a medication already in use in clinical trials.
Mitochondria, the microscopic parts found outside the nucleus of the cell that produce a cell's energy, are central players in mediating neuronal stress relevant to the pathogenesis or development of neurodegenerative diseases such as Alzheimer's disease. Mitochondrial dysfunction, or a problem with the cellular exchange of energy, is an early event observed in Alzheimer's disease. Recent studies have provided substantial evidence that mitochondria serve as direct targets for amyloid beta (Aβ) protein mediated neuronal toxicity. The observations that Aβ progressively accumulates in cortical mitochondria from Alzheimer's disease patients and in brains from transgenic Alzheimer's disease type mouse models suggest the role of mitochondrial Aβ in the pathogenesis or development of the disease. This Nature Medicine study describes how this mitochondrial process may be linked to synaptic failure in Alzheimer's disease.
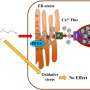
The study provides new insights into the mechanism underlying mitochondrial Aβ-mediated and synaptic stress that links to the mitochondrial permeability transition pore (mPTP), an opening that leads to cell death for those with Alzheimer's. Mitochondrial permeability transition pore causes mitochondrial swelling, outer membrane rupture and release of cell death mediators and enhances production of reactive oxygen species (ROS). Cyclophilin D (CypD), a type of enzyme called a prolyl isomerase that is located within the mitochondrial matrix, is an integral part in the formation of the mitochondrial permeability transition pore (mPTP), leading to cell death. Up until now, however, the role of CypD in Alzheimer's disease has not been elucidated.
In this paper, Dr. Yan and colleagues demonstrate that CypD interacts with Aβ peptide within the mitochondria of Alzheimer's disease patients and a transgenic mouse model of Alzheimer's disease. The cortical mitochondria isolated from Alzheimer's disease mice lacking CypD are resistant to Aβ- and Ca2+-induced mitochondria swelling and permeability transition, increase calcium buffering capacity, and attenuate generation of mitochondrial ROS. Furthermore, CypD-deficient neurons protect against Aβ- and oxidative stress-induced cell death. Importantly, deficiency of CycD greatly improved the learning, memory, and synaptic function of an Alzheimer's disease mouse model and alleviated Aβ-mediated reduction of long term potentiation (LTP). Thus, the CypD/Aβ-mediated mitochondrial permeability transition pore directly links to the cellular and synaptic perturbation relevant to the pathogenesis of Alzheimer's disease.
Source: Columbia University Medical Center