Bioinformaticians get rid of an unnecessary step in protein stability analysis
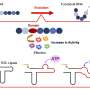
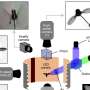
 by the structure of the original protein; (B) by the structure of its homolog; (C) by the structure of the original protein predicted based on the structure of the holomlog, and (D) by the structure predicted by artificial intelligence based on the amino acid sequence. Credit: Skolkovo Institute of Science and Technology")
Researchers from the Skoltech Center for Molecular and Cellular Biology compared different protein structure prediction methods in terms of mutant protein stability evaluation and obtained the same result for the AI-predicted structures and experimental three-dimensional (3D) of proteins with similar amino acid sequences. However, the attempt to predict the targeted protein's structure from the known structure of its "relative" only made the prediction less accurate. The team's findings will facilitate preliminary calculations in the evaluation of stability changes caused by mutation. The research was published in Bioinformatics.
Biological experiments often involve mutant proteins, which are needed for the study of the protein structure and functions or cell processes, as well as protein engineering. Mutations are known to affect a protein's structure and stability. Since experiments are too costly and time-consuming, scientists are creating a workaround in the form of computational methods to evaluate the impact of mutations on stability. However, their applications require the knowledge of a protein's 3D structure.
An experimental 3D structure is not available for all proteins and is likely to be missing for the one targeted by the team. If this is the case, 3D models of the protein's homologues, i.e. its "closest relatives," can provide the lifeline, because the degree of similarity in amino acid sequences that ensures a good match between the proteins' 3D structures is well known. The solution would be to first predict the protein's structure based on the known structure of its homologue and then calculate the impact of mutations for the predicted model.
Thanks to last year's breakthrough in protein structure prediction, the scientists now have an alternative: instead of predicting the 3D structure based on homologues, they can use the AI-based AlphaFold tool which predicts the protein structure from the amino acid sequence and has already dealt with the vast majority of proteins known to date.
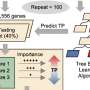
In their recent study, the Skoltech researchers decided to find out which of these approaches works best for predicting stability changes upon mutation. However accurate AlphaFold may be, finding the protein structure through experiments still remains the "gold standard." When comparing the two approaches, the team used seven stability evaluation methods and compared their results to those of AlphaFold and I-Tasser, the best homologue-based structure prediction system. Also, the researchers checked whether they can skip the homologue-based structure prediction and calculate stability for the known structure of the homologous protein.
"We decided to find out how far we would diverge from accurate prediction if we used the 'neighboring' protein structure instead of the real one. It turned out that the homology-based prediction step only makes things worse by yielding a less accurate result. We have shown that it makes virtually no difference whether you use the homologue's experimental structure or AlphaFold's prediction. In a sense, this was about validation: when faced with a new method, you have to check if it works for your task in the first place. That is exactly what we did," first author of the study, Skoltech Ph.D. student Marina Pak, comments.
"With all this fuss over AlphaFold, some scientists and non-professionals believe that it has solved all protein research issues in computational biology, but it has not. For example, the prediction of mutation-induced stability changes still displays rather low reliability, even though the change in stability is among the key drivers of protein functionality. A tool that could unambiguously determine the impact of mutation on stability would help both in planning the experiment and interpreting the results. Suppose that for a protein that is not optimal in terms of stability, we wish to find mutations that would make it stable under the desired conditions, for example, ensure that it remains active at high temperature. Once we can do this through computations alone, the approach to protein redesign and optimization will change dramatically," lead author of the study, Skoltech assistant professor Dmitry Ivankov concludes.
Although stability change prediction appears easier than predicting the 3D structure, it still remains an intractable challenge even for AI. Scarce training data is only one of the problems: AlphaFold had nearly 200,000 protein structures to train, but experimental data on stability changes amount to thousands of sets while covering a few dozen unique proteins only. The authors hope that if more data becomes available and researchers show a greater interest in the task, a breakthrough is sure to happen soon.
More information: Marina A Pak et al, Best templates outperform homology models in predicting the impact of mutations on protein stability, Bioinformatics (2022). DOI: 10.1093/bioinformatics/btac515
Journal information: Bioinformatics
Provided by Skolkovo Institute of Science and Technology