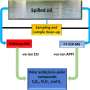
. To enable wide use of the tool, we provide both a modern web-based graphical user interface (upper panel) as well as a command line interface that enables high-performance computing (HPC) cluster usage. Credit: ISME Communications (2022). DOI: 10.1038/s43705-022-00137-0")
Schematic illustration of the glaDIAtor workflow and software. glaDIAtor is an open-source software package that implements a complete workflow for DIA metaproteomics data from raw mass spectrometry files to peptide quantifications and their taxonomic and functional annotations (lower panel). To enable wide use of the tool, we provide both a modern web-based graphical user interface (upper panel) as well as a command line interface that enables high-performance computing (HPC) cluster usage. Credit: ISME Communications (2022). DOI: 10.1038/s43705-022-00137-0
A research group from Turku Bioscience Center, Finland, has developed a new method for studying the functionality of microbiota through metaproteomics. The new method shows broad potential for the study of microbiota on a new, functional level. The characterization of the functionality of gut microbiota is central in the study of human health and disease as well as disease prediction, prevention, and treatment. Previous studies have mainly focused on cataloging the composition of microbiota, but little is known about the functionality of the human gut microbiota.
Proteins are essential for the vital functions of the body. They manage most of the cell functions and enable complex interactions between the cell and its environment. The study of proteins can therefore offer extensive information about the different functions of cells. Protein analyzes can be utilized broadly in medical research, including gut microbiome profiling.
The important role of gut microbiota in human health and their role in different diseases has been recognized in recent published studies. A research group from the University of Turku led by Professor Laura Elo has developed a new mass spectrometry-based method, which enables extensive studying of protein levels in complex microbiota samples.
"Until recently, the research on microbiota has strongly focused on discovering which microbes are present in a sample, but analyzing the functionality of the microbiota has been challenging. Recent technological advancements have however also enabled a deeper dive into the functionalities. The study of the protein levels in microbiota samples is one such rising research field, making it possible for us to reach a broader understanding of the functionality and dynamics of microbiota," says Elo.
The recently developed method utilizes newest mass spectrometry technology and advanced computational methods which enable significantly better coverage and reproducibility of the results as opposed to previous methods.
"The new method we have developed for analyzing complex protein data produces more reliable results than previous methods," says Postdoctoral Researcher Sami Pietilä.
"The currently used research methods typically only analyze the most abundant proteins, which causes fluctuation in the results from one analysis to another. The new method analyzes the samples systematically and produces reliable results without this type of fluctuation," Pietilä continues.
The computational method has been published as open source software and is freely accessible for the research community.
"It has been extremely important for us to bring this newly developed method available for all researchers as an easy-to-use application. We are also prepared to engage in further development and maintenance of this tool," adds Postdoctoral Researcher Tomi Suomi.
The study was published in ISME Communications.
More information: Sami Pietilä et al, Introducing untargeted data-independent acquisition for metaproteomics of complex microbial samples, ISME Communications (2022). DOI: 10.1038/s43705-022-00137-0
Provided by University of Turku