TSUBAME supercomputer predicts cell-membrane permeability of cyclic peptides
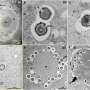
 and the predicted value of membrane permeability (vertical axis). The scatter plot on the right shows the correlation between the experimental value of membrane permeability (horizontal axis) and the value predicted by the proposed method (vertical axis). Credit: Sugita M, et al. Published by American Chemical Society (Licensed under CC BY 4.0).")
Scientists at Tokyo Institute of Technology have developed a computational method based on large-scale molecular dynamics simulations to predict the cell-membrane permeability of cyclic peptides using a supercomputer. Their protocol has exhibited promising accuracy and may become a useful tool for the design and discovery of cyclic peptide drugs, which could help us reach new therapeutic targets inside cells beyond the capabilities of conventional small-molecule drugs or antibody-based drugs.
Cyclic peptide drugs have attracted the attention of major pharmaceutical companies around the world as promising alternatives to conventional small molecule-based drugs. Through proper design, cyclic peptides can be tailored to reach specific targets inside cells, such as protein–protein interactions, which are beyond the scope of small molecules. Unfortunately, it has proven notoriously difficult to design cyclic peptides with high cell-membrane permeability—that is, cyclic peptides that can easily diffuse through the lipid bilayer that delimits the inside and outside of a cell.
In an effort to resolve this bottleneck, scientists at the Middle Molecule IT-based Drug Discovery Laboratory (MIDL) have been working on a computational method for predicting cell-membrane permeability. Established in September 2017, MIDL is one of the "Research Initiatives" at Tokyo Institute of Technology (Tokyo Tech) that goes beyond the boundaries of departments. Under the support of the Program for Building Regional Innovation Ecosystems of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), MIDL has been working with the city of Kawasaki to industrialize a framework for discovering middle molecule-based drugs—cyclic peptide drugs and nucleic acid drugs larger than conventional small-molecule drugs but smaller than antibody-based drugs—by combining computational drug design and chemical synthesis technology.
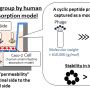
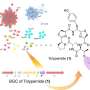
In a recent study published in the Journal of Chemical Information and Modeling, Professor Yutaka Akiyama and colleagues from MIDL and Tokyo Tech have developed a protocol for predicting the cell-membrane permeability of cyclic peptides using molecular dynamics simulations. Such simulations constitute a widely accepted computational approach for predicting and reproducing the dynamics of atoms and molecules by sequentially solving Newton's laws of motion at short time intervals. However, even a single simulation for predicting the permeability of a cyclic peptide with only eight amino acids takes a tremendous amount of time and resources. "Our study marks the first time comprehensive simulations were performed for as many as 156 different cyclic peptides," highlights Prof. Akiyama, "The simulation of each cyclic peptide using the protocol we developed took about 70 hours per peptide using 28 GPUs on the TSUBAME 3.0 supercomputer at Tokyo Tech."
The researchers verified the predicted permeability values with experimentally derived ones and confirmed an acceptable correlation coefficient of R = 0.63 under the best conditions, showcasing the potential of their protocol. Moreover, after a detailed analysis of the peptide conformation and energy values obtained from the trajectory data, Prof. Akiyama's team found that the strength of the electrostatic interactions between the atoms constituting the cyclic peptide and the surrounding media, namely lipid membrane and water molecules, are strongly related to the membrane permeability value. The simulations also revealed the way in which peptides permeate through the membrane by changing their orientation and conformation according to their surroundings (Figure 1). "Our results shed some light on the mechanisms of cell-membrane permeability and provide a guideline for designing molecules that can get inside cells more efficiently. This will greatly contribute to the development of next-generation peptide drugs," remarks Prof. Masatake Sugita, the first author of the study.
The researchers are already working on a more advanced simulation protocol that will enable more accurate predictions. They are also trying to incorporate artificial intelligence into the picture by adopting deep learning techniques, which could increase both accuracy and speed. Considering that cyclic peptides could unlock many therapeutic targets for diseases that are difficult to treat, let us hope that scientists at MIDL and Tokyo Tech succeed in their endeavors!
This research achievement will be featured in the supplementary cover of the journal issue in which this manuscript will be published.
More information: Masatake Sugita et al, Large-Scale Membrane Permeability Prediction of Cyclic Peptides Crossing a Lipid Bilayer Based on Enhanced Sampling Molecular Dynamics Simulations, Journal of Chemical Information and Modeling (2021). DOI: 10.1021/acs.jcim.1c00380
Journal information: Journal of Chemical Information and Modeling
Provided by Tokyo Institute of Technology