New model can predict multiple RNA modifications simultaneously

The ability to predict and interpret modifications of ribonucleic acid (RNA) has been a welcome advance in biochemistry research.
However, existing predictive approaches have a key drawback—they can only predict a single type of RNA modification without supporting multiple types or providing insightful interpretation of their prediction results.
Researchers from Xi'an Jiaotong-Liverpool University, led by Dr. Jia Meng, have addressed this issue by developing a model that supports 12 RNA modification types, greatly expanding RNA research prediction and interpretation.
"To the best of our knowledge, these 12 are the only widely occurring RNA modifications that can be profiled transcriptome-wide with existing base-resolution technologies. This makes them highly desirable for reliable large-scale prediction," Dr. Meng said.
Transcriptomes are the set of all RNA transcripts in a cell. By analyzing these sequences, researchers can understand which genes are turned 'on' or 'off' in the cells and related tissues.
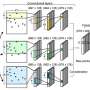
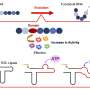
The research proved the efficacy of their MultiRM model—described by Dr. Meng as an attention-based multi-label neural network approach for integrated prediction and interpretation of RNA modifications from the primary RNA sequence. Attention mechanisms weigh the contributions of inputs to optimize the process of learning target results.
The study states that the primary purpose of the MultiRM research is to establish an interpretable predictor that could achieve state-of-the-art accuracy when identifying these RNA modifications and primary RNA sequences.
The prediction method helps researchers understand the sequence-dependent mechanisms of RNA modification, cuts wet-lab experiment costs and provides insights into the regulatory circuit of RNA metabolism.
The approach is still primarily for fundamental research only, Dr. Meng said. The tool, however, may help scientists design more efficient RNA therapeutics.
"It is hard to predict which diseases will benefit from the research, but studies indicate the enzymes of m6A RNA methylation play a key role in leukemia, lung cancer and breast cancer," he said.
To get the multiple, simultaneous predictions, the researchers used a multitask-learning framework that integrates the prediction tasks for all the 12 RNA modifications into a single prediction task, Dr. Meng said.
"Existing tools focused on the interpretation of the model," he said. "The MultiRM model provides a more comprehensive view of the epitranscriptomes and discovers underlying mechanisms of the prediction results."
The study discovered a surprising finding—the RNA modifications show significant positive associations among each other, including those originating from different nucleotides. This suggests regions exist that are intensively modified by multiple RNA modifications, which are likely to be the key regulatory components for the epitranscriptome layer of gene regulation.
How MultiRM was built
MultiRM uses a deep-learning framework on the TensorFlow platform. The researchers' approach accommodates the shared structure of different modifications while fully exploiting their distinct features.
As some modifications are more abundant than others, additional algorithms were used to balance the training data issue in multi-label learning. The researchers then implemented other machine-learning algorithms to create MultiRM's baseline benchmark.
The MultiRM model has implications for other researchers. The research team developed a web server that was designed and made accessible to serve the research community.
Researchers can freely download the data, code, and model. It takes as input an RNA sequence and returns the predicted RNA modification sites and the key sequence contents that drive the positive predictions.
More information: Zitao Song et al, Attention-based multi-label neural networks for integrated prediction and interpretation of twelve widely occurring RNA modifications, Nature Communications (2021). DOI: 10.1038/s41467-021-24313-3
MultiRM: www.xjtlu.edu.cn/biologicalsciences/multirm
Journal information: Nature Communications
Provided by Xi'an jiaotong-Liverpool University