Deep machine learning completes information about one million bioactive molecules
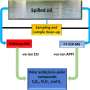
 to infer missing signatures in each bioactivity space. The inferred signatures are finally evaluated. b Coverage of the experimental version of the CC. The bar plot indicates the number of molecules available for each CC data type. The heatmap shows the cross-coverage between data sets, i.e., it is a 25 × 25 matrix capturing the proportion of molecules in one data set (rows) that are also available in other data sets (columns) c Accuracy of the 25 signaturizers, measured as the proportion of correctly classified cases within a triplet. Train–test refers to the case where the anchor molecule belongs to the test set, and the positive and negative molecules belong to the training set. Test–test corresponds to the most difficult case where none of the three molecules within the triplet has been utilized during the training. d Performance of the 25 signaturizers, measured for each molecule as the correlation between the true and predicted signatures along the 128 dimensions. Given the bimodal distribution of signature values, signatures are binarized (positive/negative) and correlation is measured as a Matthew’s correlation coefficient (MCC) over the true-vs-predicted contingency table. e Three exemplary molecules (1, 2, and 3) are shown for the D1 and E3 spaces. True and predicted signatures are displayed as color bars, both sorted according to true signature values. f Correspondingly, t-SNE 2D projections of D1 and E3 predictions, where 1, 2, and 3 are highlighted, the intensity level describes the density of molecules in the 2D space going from dark red (low density) to white (high density). g 2D-projected train (gray) and test (colored) samples for the 25 CC spaces. The legend at the bottom specifies the A1-E5 organization of the CC. From: Bioactivity descriptors for uncharacterized chemical compounds")
The Structural Bioinformatics and Network Biology laboratory, led by ICREA Researcher Dr. Patrick Aloy, has completed the bioactivity information for a million molecules using deep machine-learning computational models. It has also disclosed a tool to predict the biological activity of any molecule, even when no experimental data are available.
This new methodology is based on the Chemical Checker, the largest database of bioactivity profiles for pseudo pharmaceuticals to date, developed by the same laboratory and published in 2020. The Chemical Checker collects information from 25 spaces of bioactivity for each molecule. These spaces are linked to the chemical structure of the molecule, the targets with which it interacts or the changes it induces at the clinical or cellular level. However, this highly detailed information about the mechanism of action is incomplete for most molecules, implying that for a particular one there may be information for one or two spaces of bioactivity but not for all 25.
With this new development, researchers integrate all the experimental information available with deep machine learning methods, so that all the activity profiles, from chemistry to clinical level, for all molecules can be completed.
"The new tool also allows us to forecast the bioactivity spaces of new molecules, and this is crucial in the drug discovery process as we can select the most suitable candidates and discard those that, for one reason or another, would not work," explains Dr. Aloy.
The software library is freely accessible to the scientific community at bioactivitysignatures.org and it will be regularly updated by the researchers as more biological activity data become available. With each update of experimental data in the Chemical Checker, artificial neural networks will also be revised to refine the estimates.
Predictions and reliability
The bioactivity data predicted by the model have a greater or lesser degree of reliability depending on various factors, including the volume of experimental data available and the characteristics of the molecule.
In addition to predicting aspects of activity at the biological level, the system developed by Dr. Aloy's team provides a measure of the degree of reliability of the prediction for each molecule. "All models are wrong, but some are useful! A measure of confidence allows us to better interpret the results and highlight which spaces of bioactivity of a molecule are accurate and in which ones an error rate can be contemplated," explains Dr. Martino Bertoni, first author of the work.
Testing the system with the IRB Barcelona compound library
To validate the tool, the researchers have searched the library of compounds at IRB Barcelona for those that could be good drug candidates to modulate the activity of a cancer-related transcription factor (SNAIL1), whose activity is almost impossible to modulate due to the direct binding of drugs (it is considered an 'undruggable' target). Of a first set of 17,000 compounds, deep machine learning models predicted characteristics (in their dynamics, interaction with target cells and proteins, etc.) for 131 that fit the target.
The ability of these compounds to degrade SNAIL1 has been confirmed experimentally and it has been observed that, for a high percentage, this degradation capacity is consistent with what the models had predicted, thus validating the system.
More information: Martino Bertoni et al, Bioactivity descriptors for uncharacterized chemical compounds, Nature Communications (2021). DOI: 10.1038/s41467-021-24150-4
Journal information: Nature Communications