The molecular imaging behind COVID-19 breakthroughs

It's fair to say that before the COVID-19 pandemic, very few non-scientists could name a viral protein. But now, millions of people around the world can name the SARS-CoV-2 spike protein, and a subset of those could probably even draw a good likeness of the spike-covered virus from memory.
These nano-sized structures have received international attention because understanding how viral and immune system molecules look, at an atomic level, is the first step toward understanding how they function. And once we know how they function, we can develop vaccines and therapeutics.
The field of science dedicated to constructing molecular maps is called structural biology, and at atomic resolution it is driven by two major technologies: X-ray crystallography and cryo-electron microscopy (cryoEM). Both of these approaches have been around for many decades, yet the rapid development and deployment of SARS-CoV-2 vaccines would have been impossible without recent technical advances, especially in the resolution of cryoEM.
A team led by Paul Adams at Lawrence Berkeley National Laboratory (Berkeley Lab) is behind one of those advances. He leads an international group that created, and continues to develop, the Phenix software suite. This is used around the world to automate key steps of constructing molecular maps, which scientists call 'structure solving."
Adams, who is also director of Berkeley Lab's Molecular Biophysics & Integrated Bioimaging Division, spoke about the software's origins and how structural biologists leapt into action to combat the pandemic.
Q. How has Phenix contributed to COVID-19 research?
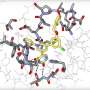
Structural biology has always played an important role in the understanding of viruses and the development of therapeutics against them. However, its role in the COVID-19 response has been critical and applied amazingly quickly. Crystallography has been used a lot to try and understand the viral proteins that actually make the virus do what it does. The structural biology community has been able to look at those proteins, their shapes, and understand how they work, as machines, and then use that information to try and figure out if there are small molecules that could potentially bind to some of these proteins and stop the virus. So that's been a major effort across the world. And, many of those structures have been analyzed using the Phenix software.
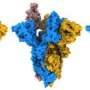
At the same time, some of the proteins in the virus aren't very easy to look at by crystallography, including the spike protein we're all so familiar with. In fact, nobody's been able to image it with crystallography. All that work has been done by electron microscopy. And so that is a place where structural information has also been crucial, because this is the thing on the outside of the virus, probably the first thing your cells see. So, in most cases, detecting the spike protein is what triggers an immune response that will hopefully end up neutralizing the viral particles. Cryo-EM using Phenix has been integral to a lot of the studies looking at how antibodies bind to the spike protein and what part of the spike protein is a good target for vaccines.
So that's how Phenix has really contributed. People were able to collect experimental data and rapidly get answers that allowed them to interpret the biology of COVID-19 infections, and pretty quickly after that, they knew what vaccines should look like. If scientists had to go off and develop a whole software program to do that analysis, it would've taken years to accomplish what they accomplished in several months.
Q. What are some examples of recent uses?
Phenix is routinely used as part of experiments on viral proteins. Some examples from the pharmaceutical company Gilead, which specializes in anti-viral therapeutics, include studies on a key hepatitis C enzyme (Bioorganic & Medicinal Chemistry Letters, Nature) and the HIV capsid protein (PNAS).
The software has also played an important role in the structural studies of coronavirus spike glycoproteins. Work on SARS-CoV and MERS (closely related coronavirus strains) used Phenix as part of their cryo-EM analyses to reveal the different structural arrangements of the spike protein, how antibodies bind to the spike protein, and to see how mutations stabilized the protein to increase antibody response. One of these mutant forms of the spike protein was used as a critical component in the development of the BioNTech vaccine [now being manufactured by Pfizer]. They used cryo-EM, and analyzed the data with Phenix, to help in the preclinical development of the BNT162b2 RNA, which has become one of the vaccines being widely used in the U.S. to fight the SARS-CoV-2 virus.
Q. How does crystallography work, and what role do computer programs play?
The first step in crystallography is to take many X-ray photographs of a crystalized version of the molecule you're interested in. These images, called diffraction patterns, tell you about the molecule's shape as they show you how X-rays are diffracted by the molecule in the crystal. Usually, hundreds or even thousands of these images that came from a synchrotron or other X-ray source need to be looked at for a particular protein crystal. That's a lot of information—often gigabytes of data. Many decades ago, people would do this by hand, poring over all the photographic prints. However, for many decades, there have been computer programs performing this first step, called data processing or data reduction.
After that data processing step, that's where Phenix steps in. It takes the information from the processed data and translates that into a map of electron densities in the crystal, which is then used to build an atomic model of the structure. These models are what scientists use to help determine a molecule's function, and what drug companies can use to help develop new therapeutics and vaccines.
Q. How did scientists perform crystallography before programs like Phenix?
Thirty years ago, many of the computer programs for structure solving were written, ad hoc, in individual labs. And so, people would have homegrown programs that were often specific to the particular structure they were trying to solve. But back then, it could take several years to solve a structure, so spending a little time coding wasn't a big deal. As crystallography technology got better, and the field of structural biology started to grow, it was clear we needed software that could make structure solving more automatic, and that could be used more broadly, so scientists didn't need to reinvent the wheel each time. That was the challenge in the field that I came from the U.K. to work on, first at Yale, then here at Berkeley Lab.
With our colleagues at Los Alamos National Laboratory, Duke University, and the University of Cambridge, we have developed Phenix over the last 20 years. It's now reached a point that it is used by thousands of groups worldwide for crystallography and increasingly cryoEM. It is great to see that Phenix has been used to help solve more than 30% of the 175,000 molecular models released in the international database (Protein Data Bank), and in the last five years it's gone up to 60% of the releases.
Nowadays, the software is very fast—often it can solve a crystal structure in less than a day. And it can now be used to solve structures from cryo-EM data, the other main structural biology technique, although the steps in Phenix can take a few days or even weeks because the structures are often larger and more complex than those in crystallography. We release updates of the software suite every six months, and we make a new version every day, so it's continually improving. We wrote Phenix with the idea that it had to be easy to use for people new to structural biology but also powerful enough for experts to solve challenging problems. This means that it is easy to download and quickly get running on an everyday computer.
Q. What has it been like to have structural biology take on so much international attention?
I will say one thing, not specific to my work with Phenix, but overall, I am hugely impressed with how much structural biology has played a role in trying to understand and combat the virus. I'm also impressed with how many structural biologists in the world quickly pivoted to work on this problem and have collaborated together. It speaks to having a great community. We really appreciate the federal and industrial funding that has made it possible to develop Phenix over the last 20 years so that it can play a role in many of these studies.
I think the past year has educated many more people about what structural biology is, and how it is a fundamental part of medicine. We've seen science deliver vaccines at an unprecedented speed, and we have some antibody therapies, but there's a lot more that can and should be done to build an arsenal against COVID-19 and future variants of the virus. So, I see a big role for structural biology and Phenix, of course, helping with that in the coming years. I think it will be years, unfortunately, but I'm really optimistic that we'll be able to crack this, and have therapeutics that address all stages of the viral life cycle.
Provided by Lawrence Berkeley National Laboratory