New efficient quantum algorithm surpasses the Quantum Phase Estimation norm

Quantum computers have seen a lot attention recently as they are expected to solve certain problems that are outside the capabilities of normal computers. Primary to these problems is determining the electronic states of atoms and molecules so they can be used more effectively in a variety of industries—from lithium-ion battery designs to in silico technologies in drug development. A common way scientists have approached this problem is by calculating the total energies of the individual states of a molecule or atom and then determine the difference in energy between these states. In nature, many molecules grow in size and complexity, and the cost to calculate this constant flux is beyond the capability of any traditional computer or currently establish quantum algorithms. Therefore, theoretical predictions of the total energies have only been possible if molecules are not sizable and isolated from their natural environment.
"For quantum computers to be a reality, its algorithms must be robust enough to accurately predict the electronic states of atoms and molecules, as they exist in nature, " state Kenji Sugisaki and Takeji Takui from the Graduate School of Science, Osaka City University.
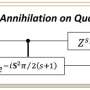
In December 2020, Sugisaki and Takui, together with their colleagues, led a team of researchers to develop a quantum algorithm they call Bayesian eXchange coupling parameter calculator with Broken-symmetry wave functions (BxB), that predicts the electronic states of atoms and molecules by directly calculating the energy differences. They noted that energy differences in atoms and molecules remain constant, regardless to how complex and large they get despite their total energies grow as the system size. "With BxB, we avoided the common practice of calculating the total energies and targeted the energy differences directly, keeping computing costs within polynomial time," they state. "Since then, our goal has been to improve the efficiency of our BxB software so it can predict the electronic sates of atoms and molecules with chemical precision."
Using the computing costs of a well-known algorithm called Quantum Phase Estimation (QPE) as a benchmark, "we calculated the vertical ionization energies of small molecules such as CO, O2, CN, F2, H2O, NH3 within 0.1 electron volts (eV) of precision," states the team, using half the number of qubits, bringing the calculation cost on par with QPE.
Their findings will be published online in the March edition of the Journal of Physical Chemistry Letters.
Ionization energy is one of the most fundamental physical properties of atoms and molecules and an important indicator for understanding the strength and properties of chemical bonds and reactions. In short, accurately predicting the ionization energy allows us to use chemicals beyond the current norm. In the past, it was necessary to calculate the energies of the neutral and ionized states, but with the BxB quantum algorithm, the ionization energy can be obtained in a single calculation without inspecting the individual total energies of the neutral and ionized states. "From numerical simulations of the quantum logic circuit in BxB, we found that the computational cost for reading out the ionization energy is constant regardless of the atomic number or the size of the molecule," the team states, "and that the ionization energy can be obtained with a high accuracy of 0.1 eV after modifying the length of the quantum logic circuit to be less than one tenth of QPE."
With the development of quantum computer hardware, Sugisaki and Takui, along with their team, are expecting the BxB quantum algorithm to perform high-precision energy calculations for large molecules that cannot be treated in real time with conventional computers.
More information: Kenji Sugisaki et al. Quantum Algorithm for the Direct Calculations of Vertical Ionization Energies, The Journal of Physical Chemistry Letters (2021). DOI: 10.1021/acs.jpclett.1c00283
Journal information: Journal of Physical Chemistry Letters
Provided by Osaka City University