Machine learning aids in simulating dynamics of interacting atoms

A revolutionary machine-learning (ML) approach to simulate the motions of atoms in materials such as aluminum is described in this week's Nature Communications journal. This automated approach to "interatomic potential development" could transform the field of computational materials discovery.
"This approach promises to be an important building block for the study of materials damage and aging from first principles," said project lead Justin Smith of Los Alamos National Laboratory. "Simulating the dynamics of interacting atoms is a cornerstone of understanding and developing new materials. Machine learning methods are providing computational scientists new tools to accurately and efficiently conduct these atomistic simulations. Machine learning models like this are designed to emulate the results of highly accurate quantum simulations, at a small fraction of the computational cost."
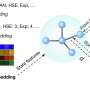
To maximize the general accuracy of these machine learning models, he said, it is essential to design a highly diverse dataset from which to train the model. A challenge is that it is not obvious, a priori, what training data will be most needed by the ML model. The team's recent work presents an automated "active learning" methodology for iteratively building a training dataset.
At each iteration, the method uses the current-best machine learning model to perform atomistic simulations; when new physical situations are encountered that are beyond the ML model's knowledge, new reference data is collected via expensive quantum simulations, and the ML model is retrained. Through this process, the active learning procedure collects data regarding many different types of atomic configurations, including a variety of crystal structures, and a variety of defect patterns appearing within crystals.
More information: Justin S. Smith et al, Automated discovery of a robust interatomic potential for aluminum, Nature Communications (2021). DOI: 10.1038/s41467-021-21376-0
Journal information: Nature Communications
Provided by Los Alamos National Laboratory