COVID-19: Future targets for treatments rapidly identified with new computer simulations

Researchers have detailed a mechanism in the distinctive corona of COVID-19 that could help scientists to rapidly find new treatments for the virus, and quickly test whether existing treatments are likely to work with mutated versions as they develop.
The team, led by the University of Warwick as part of the EUTOPIA community of European universities, have simulated movements in nearly 300 protein structures of the COVID-19 virus spike protein by using computational modelling techniques, in an effort to help identify promising drug targets for the virus.
In a new paper published today in the journal Scientific Reports, the team of physicists and life scientists detail the methods they used to model the flexibility and dynamics of all 287 protein structures for the COVID-19 virus, also known as SARS-CoV-2, identified so far. Just like organisms, viruses are composed of proteins, large biomolecules that perform a variety of functions. The scientists believe that one method for treating the virus could be interfering with the mobility of those proteins.
They have made their data, movies and structural information, detailing how the proteins move and how they deform, for all 287 protein structures for COVID-19 that were available at the time of the study, publicly accessible to allow others to investigate potential avenues for treatments.

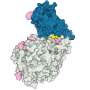
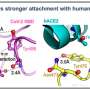
The researchers focused particular efforts on a part of the virus known as the spike protein, also called the COVID-19 echo domain structure, which forms the extended corona that gives coronaviruses their name. This spike is what allows the virus to attach itself to the ACE2 enzyme in human cell membranes, through which it causes the symptoms of COVID-19.
The spike protein is in fact a homotrimer, or three of the same type of protein combined. By modelling the movements of the proteins in the spike, the researchers identified a 'hinge' mechanism that allows the spike to hook onto a cell, and also opens up a tunnel in the virus that is a likely means of delivering the infection to the hooked cell. The scientists suggest that by finding a suitable molecule to block the mechanism—literally, by inserting a suitably sized and shaped molecule—pharmaceutical scientists will be able to quickly identify existing drugs that could be effective against the virus.
Lead author Professor Rudolf Roemer from the Department of Physics at the University of Warwick, who conducted the work while on a sabbatical at CY Cergy-Paris Université, said: "Knowing how this mechanism works is one way in which you can stop the virus, and in our study we are the first to see the detailed movement of opening. Now that you know what the range of this movement is, you can figure out what can block it.

"All those people who are interested in checking whether the protein structures in the virus could be drug targets should be able to examine this and see if the dynamics that we compute are useful to them.
"We couldn't look closely at all the 287 proteins though in the time available. People should use the motion that we observe as a starting point for their own development of drug targets. If you find an interesting motion for a particular protein structure in our data, you can use that as the basis for further modelling or experimental studies."
To investigate the proteins' movements, the scientists used a protein flexibility modelling approach. This involves recreating the protein structure as a computer model then simulating how that structure would move by treating the protein as a material consisting of solid and elastic subunits, with possible movement of these subunits defined by chemical bonds. The method has been shown to be particularly efficient and accurate when applied to large proteins such as the coronavirus's spike protein. This can allow scientists to swiftly identify promising targets for drugs for further investigation.
The protein structures that the researchers based their modelling on are all contained in the Protein Data Bank. Anyone who publishes a biological structure has to submit it to the protein databank so that it is freely available in a standard format for others to download and study further. Since the start of the COVID-19 pandemic, scientists all over the world have already submitted thousands of protein structures of COVID-19-related proteins onto the Protein Data Bank.
Professor Roemer adds: "The gold standard in modelling protein dynamics computationally is a method called molecular dynamics. Unfortunately, this method can become very time consuming particularly for large proteins such as the COVID-19 spike, which has nearly 3000 residues—the basic building blocks of all proteins. Our method is much quicker, but naturally we have to make more stringent simplifying assumptions. Nevertheless, we can rapidly simulate structures that are much larger than what alternative methods can do.
"At the moment, no-one has published experiments that identify protein crystal structures for the new variants of COVID-19. If new structures come out for the mutations in the virus then scientists could quickly test existing treatments and see if the new mechanics have an impact on their effectiveness using our method."
More information: Flexibility and mobility of SARS-CoV-2-related protein structures, Scientific Reports (2021). DOI: 10.1038/s41598-021-82849-2
Researchers have publicly released data on all protein structures to aid efforts to find potential drug targets: warwick.ac.uk/flex-covid19-data
Journal information: Scientific Reports
Provided by University of Warwick