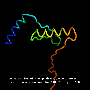Developing an AI solution to 50-year-old protein challenge

In a major scientific advance, the latest version of DeepMind's AI system AlphaFold has been recognized as a solution to the 50-year-old grand challenge of protein structure prediction, often referred to as the 'protein folding problem', according to a rigorous independent assessment. This breakthrough could significantly accelerate biological research over the long term, unlocking new possibilities in disease understanding and drug discovery among other fields.
Results from CASP14 show that DeepMind's latest AlphaFold system achieves unparalleled levels of accuracy in structure prediction. The system is able to determine highly-accurate structures in a matter of days. CASP, the Critical Assessment of protein Structure Prediction, is a biennial community-run assessment started in 1994, and the gold standard for assessing predictive techniques. Participants must blindly predict the structure of proteins that have only recently—or in some cases not yet—been experimentally determined, and wait for their predictions to be compared to experimental data.
CASP uses the "Global Distance Test (GDT)" metric to assess accuracy, ranging from 0-100. The new AlphaFold system achieves a median score of 92.4 GDT overall across all targets. The system's average error is approximately 1.6 Angstroms—about the width of an atom. According to Professor John Moult, Co-founder and Chair of CASP, a score of around 90 GDT is informally considered to be competitive with results obtained from experimental methods.
Professor John Moult, Co-Founder and Chair of CASP, University of Maryland said: "We have been stuck on this one problem—how do proteins fold up—for nearly 50 years. To see DeepMind produce a solution for this, having worked personally on this problem for so long and after so many stops and starts wondering if we'd ever get there, is a very special moment."
Why protein structure prediction matters
Proteins are essential to life and their shapes are closely linked with their functions. The ability to predict protein structures accurately enables a better understanding of what they do and how they work. There are currently over 200 million proteins in the main database and only a fraction of their 3-D structures have been mapped out.
A major challenge is the astronomical number of ways a protein could theoretically fold before settling into its final 3-D structure. Many of the greatest challenges facing society, like developing treatments for diseases or finding enzymes that break down industrial waste, are fundamentally tied to proteins and the role they play. Determining protein shapes and functions is a major field of scientific research, primarily using experimental techniques that can take years of painstaking and laborious work per structure, and require the use of multi-million dollar specialized equipment.
DeepMind's approach to the protein folding problem
This breakthrough builds on DeepMind's first entry at CASP13 in 2018, where the initial version of AlphaFold achieved the highest level of accuracy among all participants. Now, DeepMind has developed new deep learning architectures for CASP14, drawing inspiration from the fields of biology, physics, and machine learning, as well as the work of many scientists in the protein folding field over the past half-century.
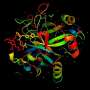
A folded protein can be thought of as a "spatial graph", where residues are the nodes and edges connect the residues in close proximity. This graph is important for understanding the physical interactions within proteins, as well as their evolutionary history. For the latest version of AlphaFold used at CASP14, DeepMind created an attention-based neural network system, trained end-to-end, that attempts to interpret the structure of this graph, while reasoning over the implicit graph that it's building. It uses evolutionarily related sequences, multiple sequence alignment (MSA), and a representation of amino acid residue pairs to refine this graph.
By iterating this process, the system develops strong predictions of the underlying physical structure of the protein. Additionally, AlphaFold can predict which parts of each predicted protein structure are reliable using an internal confidence measure.
The system was trained on publicly available data consisting of ~170,000 protein structures from the protein data bank, using a relatively modest amount of compute by modern machine learning standards—approximately 128 TPUv3-cores (roughly equivalent to ~100-200 GPUs) run over a few weeks.
Potential for real world impact
DeepMind is excited to collaborate with others to learn more about AlphaFold's potential, and the AlphaFold team is looking into how protein structure predictions could contribute to understanding of certain diseases with a few specialist groups.
There are also signs that protein structure prediction could be useful in future pandemic response efforts, as one of many tools developed by the scientific community. Earlier this year, DeepMind predicted several protein structures of the SARS-CoV-2 virus, and impressively quick work by experimentalists has now confirmed that AlphaFold achieved a high degree of accuracy on its predictions.
AlphaFold is one of DeepMind's most significant advances to date. But as with all scientific research, there's still much to be done, including figuring out how multiple proteins form complexes, how they interact with DNA, RNA, or small molecules, and how to determine the precise location of all amino acid side chains.
As with its earlier CASP13 AlphaFold system, DeepMind is planning to submit a paper detailing the workings of this system to a peer-reviewed journal in due course, and is simultaneously exploring how best to provide broader access to the system in a scalable way.
AlphaFold breaks new ground in demonstrating the stunning potential for AI as a tool to aid fundamental scientific discovery. DeepMind looks forward to collaborating with others to unlock that potential.
Professor Venki Ramakrishnan, Nobel Laureate and President of the Royal Society said: "This computational work represents a stunning advance on the protein-folding problem, a 50-year old grand challenge in biology. It has occurred decades before many people in the field would have predicted. It will be exciting to see the many ways in which it will fundamentally change biological research."
More information: deepmind.com/blog/article/alph … challenge-in-biology
Provided by DeepMind