Credit: Wyss Institute at Harvard University
DNA and RNA have been compared to "instruction manuals" containing the information needed for living "machines" to operate. But while electronic machines like computers and robots are designed from the ground up to serve a specific purpose, biological organisms are governed by a much messier, more complex set of functions that lack the predictability of binary code. Inventing new solutions to biological problems requires teasing apart seemingly intractable variables—a task that is daunting to even the most intrepid human brains.
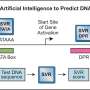
Two teams of scientists from the Wyss Institute at Harvard University and the Massachusetts Institute of Technology have devised pathways around this roadblock by going beyond human brains; they developed a set of machine learning algorithms that can analyze reams of RNA-based "toehold" sequences and predict which ones will be most effective at sensing and responding to a desired target sequence. As reported in two papers published concurrently today in Nature Communications, the algorithms could be generalizable to other problems in synthetic biology as well, and could accelerate the development of biotechnology tools to improve science and medicine and help save lives.
"These achievements are exciting because they mark the starting point of our ability to ask better questions about the fundamental principles of RNA folding, which we need to know in order to achieve meaningful discoveries and build useful biological technologies," said Luis Soenksen, Ph.D., a Postdoctoral Fellow at the Wyss Institute and Venture Builder at MIT's Jameel Clinic who is a co-first author of the first of the two papers.
Getting ahold of toehold switches
The collaboration between data scientists from the Wyss Institute's Predictive BioAnalytics Initiative and synthetic biologists in Wyss Core Faculty member Jim Collins' lab at MIT was created to apply the computational power of machine learning, neural networks, and other algorithmic architectures to complex problems in biology that have so far defied resolution. As a proving ground for their approach, the two teams focused on a specific class of engineered RNA molecules: toehold switches, which are folded into a hairpin-like shape in their "off" state. When a complementary RNA strand binds to a "trigger" sequence trailing from one end of the hairpin, the toehold switch unfolds into its "on" state and exposes sequences that were previously hidden within the hairpin, allowing ribosomes to bind to and translate a downstream gene into protein molecules. This precise control over the expression of genes in response to the presence of a given molecule makes toehold switches very powerful components for sensing substances in the environment, detecting disease, and other purposes.

Credit: Wyss Institute at Harvard University
However, many toehold switches do not work very well when tested experimentally, even though they have been engineered to produce a desired output in response to a given input based on known RNA folding rules. Recognizing this problem, the teams decided to use machine learning to analyze a large volume of toehold switch sequences and use insights from that analysis to more accurately predict which toeholds reliably perform their intended tasks, which would allow researchers to quickly identify high-quality toeholds for various experiments.
The first hurdle they faced was that there was no dataset of toehold switch sequences large enough for deep learning techniques to analyze effectively. The authors took it upon themselves to generate a dataset that would be useful to train such models. "We designed and synthesized a massive library of toehold switches, nearly 100,000 in total, by systematically sampling short trigger regions along the entire genomes of 23 viruses and 906 human transcription factors," said Alex Garruss, a Harvard graduate student working at the Wyss Institute who is a co-first author of the first paper. "The unprecedented scale of this dataset enables the use of advanced machine learning techniques for identifying and understanding useful switches for immediate downstream applications and future design."
Armed with enough data, the teams first employed tools traditionally used for analyzing synthetic RNA molecules to see if they could accurately predict the behavior of toehold switches now that there were manifold more examples available. However, none of the methods they tried—including mechanistic modeling based on thermodynamics and physical features—were able to predict with sufficient accuracy which toeholds functioned better.
A picture is worth a thousand base pairs
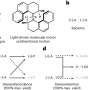
The researchers then explored various machine learning techniques to see if they could create models with better predictive abilities. The authors of the first paper decided to analyze toehold switches not as sequences of bases, but rather as two-dimensional "images" of base-pair possibilities. "We know the baseline rules for how an RNA molecule's base pairs bond with each other, but molecules are wiggly—they never have a single perfect shape, but rather a probability of different shapes they could be in," said Nicolaas Angenent-Mari, a MIT graduate student working at the Wyss Institute and co-first author of the first paper. "Computer vision algorithms have become very good at analyzing images, so we created a picture-like representation of all the possible folding states of each toehold switch, and trained a machine learning algorithm on those pictures so it could recognize the subtle patterns indicating whether a given picture would be a good or a bad toehold."
Another benefit of their visually-based approach is that the team was able to "see" which parts of a toehold switch sequence the algorithm "paid attention" to the most when determining whether a given sequence was "good" or "bad." They named this interpretation approach Visualizing Secondary Structure Saliency Maps, or VIS4Map, and applied it to their entire toehold switch dataset. VIS4Map successfully identified physical elements of the toehold switches that influenced their performance, and allowed the researchers to conclude that toeholds with more potentially competing internal structures were "leakier" and thus of lower quality than those with fewer such structures, providing insight into RNA folding mechanisms that had not been discovered using traditional analysis techniques.
"Being able to understand and explain why certain tools work or don't work has been a secondary goal within the artificial intelligence community for some time, but interpretability needs to be at the forefront of our concerns when studying biology because the underlying reasons for those systems' behaviors often cannot be intuited," said Jim Collins, Ph.D., the senior author of the first paper. "Meaningful discoveries and disruptions are the result of deep understanding of how nature works, and this project demonstrates that machine learning, when properly designed and applied, can greatly enhance our ability to gain important insights about biological systems." Collins is also the Termeer Professor of Medical Engineering and Science at MIT.
Now you're speaking my language
While the first team analyzed toehold switch sequences as 2-D images to predict their quality, the second team created two different deep learning architectures that approached the challenge using orthogonal techniques. They then went beyond predicting toehold quality and used their models to optimize and redesign poorly performing toehold switches for different purposes, which they report in the second paper.
The first model, based on a convolutional neural network (CNN) and multi-layer perceptron (MLP), treats toehold sequences as 1D images, or lines of nucleotide bases, and identifies patterns of bases and potential interactions between those bases to predict good and bad toeholds. The team used this model to create an optimization method called STORM (Sequence-based Toehold Optimization and Redesign Model), which allows for complete redesign of a toehold sequence from the ground up. This "blank slate" tool is optimal for generating novel toehold switches to perform a specific function as part of a synthetic genetic circuit, enabling the creation of complex biological tools.
"The really cool part about STORM and the model underlying it is that after seeding it with input data from the first paper, we were able to fine-tune the model with only 168 samples and use the improved model to optimize toehold switches. That calls into question the prevailing assumption that you need to generate massive datasets every time you want to apply a machine learning algorithm to a new problem, and suggests that deep learning is potentially more applicable for synthetic biologists than we thought," said co-first author Jackie Valeri, a graduate student at MIT and the Wyss Institute.

Credit: Wyss Institute at Harvard University
The second model is based on natural language processing (NLP), and treats each toehold sequence as a "phrase" consisting of patterns of "words," eventually learning how certain words are put together to make a coherent phrase. "I like to think of each toehold switch as a haiku poem: like a haiku, it's a very specific arrangement of phrases within its parent language—in this case, RNA. We are essentially training this model to learn how to write a good haiku by feeding it lots and lots of examples," said co-first author Pradeep Ramesh, Ph.D., a Visiting Postdoctoral Fellow at the Wyss Institute and Machine Learning Scientist at Sherlock Biosciences.
Ramesh and his co-authors integrated this NLP-based model with the CNN-based model to create NuSpeak (Nucleic Acid Speech), an optimization approach that allowed them to redesign the last 9 nucleotides of a given toehold switch while keeping the remaining 21 nucleotides intact. This technique allows for the creation of toeholds that are designed to detect the presence of specific pathogenic RNA sequences, and could be used to develop new diagnostic tests.
The team experimentally validated both of these platforms by optimizing toehold switches designed to sense fragments from the SARS-CoV-2 viral genome. NuSpeak improved the sensors' performances by an average of 160%, while STORM created better versions of four "bad" SARS-CoV-2 viral RNA sensors whose performances improved by up to 28 times.
"A real benefit of the STORM and NuSpeak platforms is that they enable you to rapidly design and optimize synthetic biology components, as we showed with the development of toehold sensors for a COVID-19 diagnostic," said co-first author Katie Collins, an undergraduate MIT student at the Wyss Institute who worked with MIT Associate Professor Timothy Lu, M.D., Ph.D., a corresponding author of the second paper.
"The data-driven approaches enabled by machine learning open the door to really valuable synergies between computer science and synthetic biology, and we're just beginning to scratch the surface," said Diogo Camacho, Ph.D., a corresponding author of the second paper who is a Senior Bioinformatics Scientist and co-lead of the Predictive BioAnalytics Initiative at the Wyss Institute. "Perhaps the most important aspect of the tools we developed in these papers is that they are generalizable to other types of RNA-based sequences such as inducible promoters and naturally occurring riboswitches, and therefore can be applied to a wide range of problems and opportunities in biotechnology and medicine."
Additional authors of the papers include Wyss Core Faculty member and Professor of Genetics at HMS George Church, Ph.D.; and Wyss and MIT Graduate Students Miguel Alcantar and Bianca Lepe.
"Artificial intelligence is wave that is just beginning to impact science and industry, and has incredible potential for helping to solve intractable problems. The breakthroughs described in these studies demonstrate the power of melding computation with synthetic biology at the bench to develop new and more powerful bioinspired technologies, in addition to leading to new insights into fundamental mechanisms of biological control," said Don Ingber, M.D., Ph.D., the Wyss Institute's Founding Director. Ingber is also the Judah Folkman Professor of Vascular Biology at Harvard Medical School and the Vascular Biology Program at Boston Children's Hospital, as well as Professor of Bioengineering at Harvard's John A. Paulson School of Engineering and Applied Sciences.
More information: "A Deep Learning Approach to Programmable RNA Switches" Nature Communications (2020).
"Sequence-to-function deep learning frameworks for engineered riboregulators" Nature Communications (2020).
Journal information: Nature Communications
Provided by Harvard University