A genome-scale map of DNA methylation kinetics

While the first genome-wide DNA methylation map in mammalian cells was established over 10 years ago, such maps only provide snapshots and do not inform about the actual dynamics of this epigenetic mark. Researchers from the Schübeler group now quantified actual rates of methylation and demethylation for 860,404 individual CpGs in mouse embryonic stem cells. Their study reveals highly variable and context-specific activity for the DNA methylation machinery.
DNA methylation is an essential epigenetic mark in eukaryotes and plays a role in gene regulation. It occurs most frequently at cytosines that are followed by guanines (CpG), where high levels of DNA methylation in promoter regions are typically associated with gene repression.
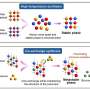
DNA methylation levels are governed by opposing enzymatic reactions that apply and remove the mark. In mammalian cells, the methylation process can be divided into two types carried out by two classes of DNA methyltransferases (DNMTs): maintenance methylation (to preserve DNA methylation after every cellular DNA replication cycle) and de novo methylation (to set up DNA methylation patterns early in development). Conversely, DNA methylation can be lost either by incomplete maintenance following replication, referred to as passive demethylation, or actively via specific enzymes called TETs. The rates of enzymatic activity eventually drive the methylation process to a steady-state, resulting in the average methylation patterns observed in various cell types.
It has been a challenge to actually assign numbers to enzyme activity rates in the context of native chromatin. Now, researchers from the Schübeler group, led by postdoc Paul Ginno, computational biologist Dimos Gaidatzis and former Ph.D. student Angelika Feldmann studied all three processes—de novo and maintenance methylation, as well as active demethylation—in mouse embryonic stem cells. They combined genetic ablations of methylating and demethylating enzymes with high-coverage quantitative measurements of DNA methylation and sophisticated modelling in order to quantify actual rates of methylation and demethylation for individual CpGs at the scale of the genome.
Paul Ginno explains: "Studying methylation as a steady-state process does not reveal actual methylation turnover. By comparing the rates of methylation/demethylation of CpGs in different genomic locations, we found that rates could differ by two orders of magnitude for CpGs with similar steady-state measurements."
Another important finding of the study—which has recently been published in Nature Communications—suggests that transcription factor binding not only hinders de novo methylation, but also represents a challenge for maintenance methylation. These results speak for a model where many transcription factors rebind DNA quickly after the replication fork, interfering with maintenance methylation.
Ginno and colleagues also discovered that methylation turnover is elevated in gene bodies (the part of the gene that is being transcribed). While gene body methylation is a highly conserved pattern, its function remains enigmatic. This study now reveals that methylation turnover increases with transcriptional activity and that the high overall methylation observed at genes is in constant flux as a function of transcription.
"It's the first time that a genome-scale map of DNA methylation kinetics has been done at this level," says group leader Dirk Schübeler. "This was a challenging study: setting up the experimental system by Angelika proved to be complex as was the solving of the data modeling part by Dimos from the Computational Biology team! A great team effort."
More information: Paul Adrian Ginno et al. A genome-scale map of DNA methylation turnover identifies site-specific dependencies of DNMT and TET activity, Nature Communications (2020). DOI: 10.1038/s41467-020-16354-x
Journal information: Nature Communications