June 29, 2020 feature
Structural evidence for a dynamic metallocofactor during dinitrogen reduction by Mo-nitrogenase
 the interface of chains A and B [P-cluster(A/B)] and (D to F) the interface of chains C and D [P-cluster(C/D)] of Av1*. Chains A and C are the a-subunits, and chains B and D are the b subunits of the two ab dimers of Av1*. [(A) and (D)] The P-clusters are shown in ball-and-stick presentation, and the key residues interacting with the P-clusters are indicated as sticks. Chains A and C are shown as wheat ribbons, and chains B and D are shown as light-blue ribbons. [(B) and (C)] P-cluster(A/B) and [(E) and (F)] P-cluster(C/D) superimposed with [(B) and (E)] the anomalous density maps calculated at 7100 eV at a resolution of 2.18 Å and contoured at 4.0s, showing the position of sulfur atoms (mint-blue mesh); and with [(C) and (F)] the anomalous density maps calculated at 7141 eV at a resolution of 2.1 Å and contoured at 15.0s, showing the position of iron atoms (red mesh). Atoms are colored as follows: Fe, orange; S, yellow; O, red; N, blue. Single-letter abbreviations for the amino acid residues are as follows: C, Cys; G, Gly; H, His; R, Arg; S, Ser. Credit: Science, doi: 10.1126/science.aaz6748")
The enzyme nitrogenase is a biological catalyst that can reduce dinitrogen (N2) to ammonia in the presence of a suite of complex metallocofactors. However, the mechanistic details of the reaction remain scarce. In a new report on Science, Wonchull Kang and a research team in chemistry, molecular biology and biochemistry at the University of California-Irvine, U.S., reported a 1.83-angstrom crystal structure for the nitrogenase molybdenum-iron (MoFe) protein, which they captured under physiological dinitrogen turnover conditions. The results of the study can assess the possible mechanisms of N2 reduction and the role of belt-sulfur sites during the process.
Nitrogenase is a catalyst for a critical step in the global nitrogen cycle, during the ambient reduction of atmospheric dinitrogen (N2) to the bioavailable ammonia (NH3). The molybdenum nitrogenase enzyme contains two protein components: one containing the iron (Fe) protein in an iron sulfur (Fe4S4) cluster with an adenosine triphosphate (ATP)-binding site within each subunit. The other, molybdenum iron (MoFe) protein containing an α2β2 heterotetramer with two complex metalloclusters. During molybdenum-nitrogenase (Mo-nitrogenase) catalysis, the repeated association and dissociation between the two protein components permitted ATP-dependent electron transfer from the Fe4S4 cluster to the MoFe protein for substrate reduction. The ability of nitrogenase to shuttle many electrons to its cofactor made the enzyme highly versatile during substrate reduction.
Understanding the mechanism-of-action of the nitrogenase enzyme
Many efforts have gone to understand the mechanisms of nitrogenase since its discovery, where some had focused on substrate and inhibitor interactions of the enzyme. Of these efforts, Kang et al. determined a strategy worthy of consideration by limiting excess electron supplies that inadvertently drove the N2 reduction process forward. This reverted the substrate or intermediate-bound state of the enzyme to a resting state or reduced the enzyme to an indiscernible mixed state. The process was relevant since nitrogenase proteins are routinely isolated in the presence of excess dithionite as an externally supplied reductant, and the removal of this artificial electron source in the absence of oxygen could help scientists to capture dinitrogen (N2) or its intermediates for analysis.
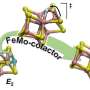
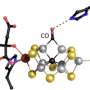
 and transparent (right) ribbon presentations of the heterotetramer of Av1*, with the a-subunits (Chain-A and Chain-C) and b-subunits (Chain-B and Chain-D) colored in wheat and blue, respectively. The M- and P-clusters are illustrated as space-filling models. Color code of atoms: Fe, orange; S, yellow; O, red; N, blue; Mo, cyan; C, gray. The M-clusters in Chain-A and Chain-C are designated M-cluster(A) and M-cluster(C), respectively. The P-clusters at the Chain-A/Chain-B and Chain-C/Chain-D interfaces are designated P-cluster(A/B) and P-cluster(C/D), respectively. PYMOL was used to prepare this figure. Credit: Science, doi: 10.1126/science.aaz6748")
As a proof of concept, Kang et al. prepared the crude extract of an anerobic bacterial strain Azotobacter vinelandii with or without the addition of dithionite after cell disruption. The A. vinelandii strain actively expressed a Mo-nitrogenase containing a histidine-tagged MoFe protein in both cases. When they analyzed the activity of these samples, the dithionite-free crude extract samples were nearly inactive during substrate reduction—due to depletion of electrons in the crude extracts during cell disruption. Kang et al. could therefore fully restore the activity of samples by adding dithionite and ATP (i.e. by supplying electrons).
Clusters of nitrogenase—two unique metalloclusters: the P-cluster and the M-cluster.
Based on the outlined conditions, when a nitrogenase-expressing culture that actively performs N2 fixation is subjected to cell lysis without additional electron supplies, the nitrogenase remained functional. Although potentially arrested in a "dormant" or intermediate-bound state due to withdrawal of electron flow into an iron-sulfur metallocluster known as the M-cluster, located within the nitrogenase enzyme. When Kang et al. purified the dithionite-free crude extract, the histidine-tagged MoFe protein (designated as AV1* in the study) was active during N2 reduction and also fully functional. When the team crystallized AV1*, they observed brown crystals that diffracted to a resolution of 1.83 angstrom (Å). They confirmed the structural rearrangement of the two P-clusters of AV1* using anomalous density data and used electron paramagnetic resonance to observe the structural assignment. The results provided them long-sought answers to the physiological relevance of this experimental state and pointed to a limited flow of electrons between the two unique metalloclusters (P- and M-clusters) of the compound in the absence of dithionite.

A plausible mechanism of action
A plausible mechanism of action that agreed with the experimental observations included the stepwise reduction of dinitrogen (N2) at the three belt sulfur sites on the nitrogenase catalyst based on the rotation of the M-cluster. The proposed mechanism begins with tight binding of N2 at a specific site, followed by rotation of the bound N2 to another subsequent site (sites designated as S3A to S2B to S5A on the compound). During the process, the reduction/protonation of N2 to the diazene level occurred through hydrogen bonding, followed by further reduction/protonation for its conversion into ammonia, prior to its release from the structure. Subsequent rotation of the cluster brought a new N2 molecule to the next site to initiate the next round of stepwise N2 reduction through continued cluster rotation in a delicate mechanism during catalysis. Such cycling between different reactions sites were loosely analogous to the mechanism of the ATP synthase enzyme. The rotating metallocluster thus effectively allowed for the multi-electron reduction of N2 through a divide-and-conquer approach.
. Structures of M-clusters in (A and B) chain A [M-cluster(A)] and (C and D) chain C [M-cluster(C)] refined at a resolution of 1.73 Å. Side view of (A) M-cluster(A) and (C) M-cluster(C) with key residues interacting with the clusters indicated as sticks. M-cluster(A) and M-cluster(C) are superimposed with the Fo-Fc omit maps of the belt sulfurs contoured at 13s (mint-blue mesh). View along the Fe1-C-Mo direction of (B) M-cluster(A) and (D) M-cluster(C) superimposed with the anomalous density maps calculated at 7100 eV at a resolution of 2.17 Å and contoured at 4.0s, showing the presence of the anomalous sulfur density (mint-blue mesh) at all belt sulfur positions (S2B, S3A, and S5A) in (B) M-cluster(A) and (D) M-cluster(C). Credit: Science, doi: 10.1126/science.aaz6748")
To understand the sulfur-displaced conformation of AV1* under limited electron flux, the team formed AV1* turnover with dithionite (designated as AV1*TOD), to yield brown crystals that diffracted to a resolution of 1.73 Å. The observations were consistent with the mechanism of bound dinitrogen species on the compound and illustrated the physiological relevance of the conformation during catalysis. The capacity to displace three different sites by a dinitrogen species was consistent with previous investigations on catalysis-dependent selenium. Kang et al. proposed many mechanisms to explain the observations, however they seek further experimental support to verify them. The team highlighted the possibility for all belt-sulfur sites to be involved in the process of catalysis due to the presence of asymmetric belt-sulfur displacements in the compound. The results aim to provoke a paradigm shift in the mechanistic thinking of nitrogenase activity, ultimately to understand the intricate mechanism of the enzyme.
More information: Wonchull Kang et al. Structural evidence for a dynamic metallocofactor during N2 reduction by Mo-nitrogenase, Science (2020). DOI: 10.1126/science.aaz6748
Douglas C Rees et al. Structural basis of biological nitrogen fixation, Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences (2005). DOI: 10.1098/rsta.2004.1539
S. C. Lee et al. Speculative synthetic chemistry and the nitrogenase problem, Proceedings of the National Academy of Sciences (2003). DOI: 10.1073/pnas.0630028100
Journal information: Science , Proceedings of the National Academy of Sciences
Provided by Science X Network
© 2020 Science X Network