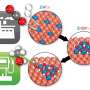
Water is key in catalytic conversion of methane to methanol

Scientists at the U.S. Department of Energy's Brookhaven National Laboratory and collaborators have revealed new details that explain how a highly selective catalyst converts methane, the main component of natural gas, to methanol, an easy-to-transport liquid fuel and feedstock for making plastics, paints, and other commodity products. The findings could aid the design of even more efficient/selective catalysts to make methane conversion an economically viable and environmentally attractive alternative to venting or flaring "waste" gas.
As described in a paper appearing in Science, the team used theory-based models and simulations to identify the atomic-level rearrangements that take place during the reaction, and then conducted experiments to verify those details. The studies revealed three essential roles for water, working in conjunction with an economical cerium-oxide/copper-oxide catalyst, to bring about the conversion of methane to methanol with 70 percent selectivity while blocking unwanted side reactions.
"We knew from previous work that we'd developed a highly selective catalyst for the direct conversion of methane to methanol in the presence of water," said Brookhaven Lab chemist Sanjaya Senanayake, who led the project. "But now, using advanced theoretical and experimental techniques, we've learned why it works so well."
The findings could speed the development of catalysts that make use of methane escaping from gas and oil wells, where it is typically vented directly into the atmosphere or burned off.
"Transporting gas is extremely difficult and potentially hazardous," Senanayake said. "But if you convert it directly into a liquid you can move it and use it instead of flaring it wastefully. While the commercialization potential for such a reaction may still take several years, we hope that our results and the understanding of how it all works will help to get there faster."
Theory lays the groundwork
The search for methane-to-methanol catalysts has turned up a few promising prospects. But many operate in several distinct steps with high energy requirements. And in many cases, competing reactions break down the methane (and any produced methanol) completely to carbon monoxide (CO) and CO2. So, when the Brookhaven team first observed that their catalyst could directly convert methane to methanol with high yield in one continuous reaction, they wanted to know more about how it performed this difficult task.
They were particularly interested in figuring out the role of water, which appeared to facilitate key steps in the process and somehow block the reaction pathways that produced CO and CO2.
Using computational tools in Brookhaven Lab's Center for Functional Nanomaterials (CFN), Brookhaven's Scientific Data and Computing Center, Stony Brook University (SBU), and the National Energy Research Scientific Computing Center (NERSC) at DOE's Lawrence Berkeley National Laboratory (Berkeley Lab), Brookhaven chemist Ping Liu developed the theoretical approach to figure out what was going on.
First, she used "density functional theory" (DFT) calculations to identify how the reactants (methane, oxygen, and water) changed as they interacted with one another and the cerium-oxide/copper-oxide catalyst at various stages during the reaction. These calculations also included information about how much energy it would take to get from one atomic arrangement to the next.
"The DFT gives you a bunch of 'snapshots' of the stages involved in the reaction and the 'bumps' or barriers you have to overcome to get from one stage to the next," she explained.
Then she performed "kinetic Monte Carlo" simulations—essentially using computers to try out all the possible ways the reaction could proceed from snapshot to snapshot. The simulations take into account all the possible pathways and energy requirements to move from one stage to the next.
"These simulations start with each intermediate stage and look at all the possibilities that can go to the next step—and figure out what is the most probable pathway," Liu said. "The simulations determine the most probable way the snapshots can be connected in real time."
The simulations also model how different reaction conditions—for example, changes in pressure and temperature—will affect reaction rates and the probable pathways.
"There were 45-50 possible components in the 'reaction network' we were simulating," said Jose Rodriguez, a leader of Brookhaven's catalysis group who also has a joint appointment at SBU. "Out of those, Ping, Erwei Huang, and Wenjie Liao, two Ph.D. students at SBU, were able to predict what would be the most favorable conditions, the best path, for going from methane to methanol and not to CO and CO2—and all induced by the presence of water."
The models predicted three roles for water: 1) activating the methane (CH4) by breaking one carbon-hydrogen bond and providing an -OH group to convert the CH3 fragment to methanol, 2) blocking reactive sites that could potentially convert methane and methanol to CO and CO2, and 3) facilitates the displacement of methanol formed on the surface into the gas phase as a product.
"All the action takes place at one or two active sites at the interface between the cerium-oxide nanoparticles and copper-oxide film that make up our catalyst," Senanayake said.
But this description was still just a model. The scientists needed evidence.
 members of the research team. First row, left to right: Sanjaya Senanayake (Brookhaven), Mausumi Mahapatra (Brookhaven), Jose A Rodriguez (Brookhaven), Ping Liu (Brookhaven) and Wenjie Liao (SBU). Second row: Ivan Orozco (SBU), Ning Rui (Brookhaven), Zongyuan Liu (Brookhaven) and Erwei Huang (SBU). Credit: Brookhaven National Laboratory")
Experiments provide proof
To gather evidence, the scientists from Brookhaven and SBU conducted additional experiments in Brookhaven's Chemistry Division laboratories and took several trips to the Advanced Light Source (ALS) at Berkeley Lab. This team included SBU Ph.D. student Ivan Orozco and post-doctoral fellows Zongyuan Liu, Robert M. Palomino, Ning Rui, and Mausumi Mahapatra.
At the ALS, the group worked with Berkeley Lab's Slavomir Nemsak and collaborators Thomas Duchon (Peter-Grünberg-Institut in Germany) and David Grinter (Diamond Light Source in the United Kingdom) to perform experiments using ambient pressure (AP) X-ray photoelectron spectroscopy (XPS), which allowed them to track the reaction as it happened in real time to identify key steps and intermediates.
"The x-rays excite electrons, and the energy of the electrons tells you what chemical species you have on the surface and the chemical state of the species. It makes a 'chemical fingerprint.'" said Rodriguez. "Using this technique, you can follow the surface chemistry and reaction mechanism in real time."
Running the reaction with and without water under a range of conditions confirmed that water played the predicted three roles. The measurements showed how the reaction conditions moved the process forward and maximized the production of methanol by preventing side reactions.
"We found direct evidence for formation of CH3O—an intermediate precursor for methanol—in the presence of water," Rodriguez said. "And because you have the water, you modify all the surface chemistry to block the side reactions, and also easily release the methanol from the catalyst surface so it doesn't decompose."
"Now that we've identified the design principles for the catalyst," Senanayake said, "next we have to build a real system for using such a catalyst and test it—and see if we can make it better."
More information: "Water-promoted interfacial pathways in methane oxidation to methanol on a CeO2-Cu2O catalyst" Science (2020). science.sciencemag.org/cgi/doi … 1126/science.aba5005
Journal information: Science
Provided by Brookhaven National Laboratory