Predicting reaction results: Machines learn chemistry

Everyday life without artificial intelligence is barely conceivable in today's world. Countless applications in areas such as autonomous driving, foreign language translation or medical diagnostics have found their way into our lives. In chemical research, too, great efforts are being made to apply artificial intelligence (AI), also known as machine learning, effectively. These technologies have already been used to predict the properties of individual molecules, making it easier for researchers to select the compound to be produced.
This production, known as synthesis, usually involves considerable effort as there are many possible synthesis routes to producing a target molecule. Since the success of each individual reaction depends on numerous parameters, it is not always possible, even for experienced chemists, to predict whether a reaction will take place—and even less how well it will work. In order to remedy this situation, a team of chemists and computer scientists from the University of Münster (Germany) has joined forces and developed an AI tool which has now been published in the journal Chem.
Background and method:
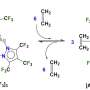
"A chemical reaction is a highly complex system," explains Frederik Sandfort, Ph.D. student at the Institute of Organic Chemistry and one of the lead authors of the publication. "In contrast to the prediction of properties of individual compounds, a reaction is the interaction of many molecules and thus a multidimensional problem," he adds. Moreover, there are no clearly defined "rules of the game" which, as in the case of modern chess computers, simplify the development of AI models. For this reason, previous approaches to accurately predicting reaction results such as yields or products are mostly based on a previously gained understanding of molecular properties. "The development of such models involves a great deal of effort. Moreover, the majority of them are highly specialized and cannot be transferred to other problems," Frederik Sandfort adds.
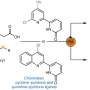
The focus of the work presented was therefore on a general applicability of the programme, so that other chemists can easily use it for their own work. To ensure this, the model is based directly on molecular structures. "Every organic compound can be represented as a graph, in principle as an image," explains Marius Kühnemund, another author, from the field of computer science. "On such graphs, simple structural queries—comparable to the question of colours or shapes in a photo—can be made in order to capture the so-called chemical environment as accurately as possible."
The combination of many such successive queries results in a so-called molecular fingerprint. These simple number sequences have long been used in chemoinformatics to find structural similarities and are well suited for computer-aided applications. In their approach, the authors use a large number of such fingerprints to represent the chemical structure of each molecule as accurately as possible. "In this way, we have been able to develop a robust system that can be used to predict completely different reaction results," adds Marius Kühnemund, "The same model can be used to predict both yields and stereoselectivities, which is unique."
The authors demonstrated that their programme can be applied easily and allows accurate predictions, especially in combination with modern robotics, by using a data set that was not originally created for machine learning. "This data set contains only relative sales of the starting materials and no exact yields," Frederik Sandfort explains. "For exact yields, calibrations have to be created. However, due to the high effort involved, this is rarely done in reality."
The team will continue to develop their programme further and equip it with new functions in the future. Prof. Frank Glorius is confident: "When it comes to evaluating large amounts of complex data, computers are fundamentally superior to us. However, our goal is not to replace synthetic chemists with machines, but to support them as effectively as possible. Models based on artificial intelligence can significantly change the way we approach chemical syntheses. But we are still at the very beginning."
More information: Frederik Sandfort et al, A Structure-Based Platform for Predicting Chemical Reactivity, Chem (2020). DOI: 10.1016/j.chempr.2020.02.017
Journal information: Chem
Provided by University of Münster