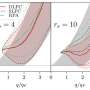
as a function of the process duration, compared to the traditional method (logarithmic scale). Credit: Niclas Schlünzen, AG Bonitz")
Computing time required for the new G1-G2 method (solid line) as a function of the process duration, compared to the traditional method (logarithmic scale). Credit: Niclas Schlünzen, AG Bonitz
How an electron behaves in an atom, or how it moves in a solid, can be predicted precisely with the equations of quantum mechanics. These theoretical calculations agree fully with the results obtained from experiments. But complex quantum systems, which contain many electrons or elementary particles—such as molecules, solids or atomic nuclei—can currently not be described exactly, even with the most powerful computers available today. The underlying mathematical equations are too complex, and the computational requirements are too large. A team led by Professor Michael Bonitz from the Institute of Theoretical Physics and Astrophysics at Kiel University (CAU) has now succeeded in developing a simulation method, which enables quantum mechanical calculations up to around 10,000 times faster than previously possible. They have published their findings in the current issue of the renowned scientific journal Physical Review Letters.
Even with extremely powerful computers, quantum simulations take too long
The new procedure of the Kiel researchers is based on one of the currently most powerful and versatile simulation techniques for quantum mechanical many-body systems. It uses the method of so-called nonequilibrium Green functions: this allows movements and complex interactions of electrons to be described with very high accuracy, even for an extended period. However, to date this method is very computer-intensive: in order to predict the development of the quantum system over a ten times longer period, a computer requires a thousand times more processing time.
With the mathematical trick of introducing an additional auxiliary variable, the physicists at the CAU have now succeeded in reformulating the primary equations of nonequilibrium Green functions such that the calculation time only increases linearly with the process duration. Thus, a ten times longer prediction period only requires ten times more computing time. In comparison with the previously-used methods, the physicists achieved an acceleration factor of approximately 10,000. This factor increases further for longer processes. Since the new approach combines two Green functions for the first time, it is called "G1-G2 method."
Temporal development of material properties predictable for the first time
The new calculation model of the Kiel research team not only saves expensive computing time, but also allows for simulations, which have previously been completely impossible. "We were surprised ourselves that this dramatic acceleration can also be demonstrated in practical applications," explained Bonitz. For example, it is now possible to predict how certain properties and effects in materials such as semiconductors develop over an extended period of time. Bonitz is convinced: "The new simulation method is applicable in numerous areas of quantum many-body theory, and will enable qualitatively new predictions, such as about the behaviour of atoms, molecules, dense plasmas and solids after excitation by intense laser radiation."
More information: Niclas Schlünzen, Jan-Philip Joost, Michael Bonitz, Achieving the Scaling Limit for Nonequilibrium Green Functions Simulations, Physical Review Letters 124, 7, (2020) DOI: 10.1103/PhysRevLett.124.076601
Journal information: Physical Review Letters
Provided by Kiel University