Researchers cast neural nets to simulate molecular motion

New work from Los Alamos National Laboratory, the University of North Carolina at Chapel Hill, and the University of Florida is showing that artificial neural nets can be trained to encode quantum mechanical laws to describe the motions of molecules, supercharging simulations potentially across a broad range of fields.
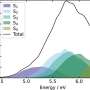
"This means we can now model materials and molecular dynamics billions of times faster compared to conventional quantum methods, while retaining the same level of accuracy," said Justin Smith, Los Alamos physicist and Metropolis Fellow in the laboratory's Theoretical Division. Understanding how molecules move is critical to tapping their potential value for drug development, protein simulations and reactive chemistry, for example, and both quantum mechanics and experimental (empirical) methods feed into the simulations.
The new technique, called the ANI-1ccx potential, promises to advance the capabilities of researchers in many fields and improve the accuracy of machine learning-based potentials in future studies of metal alloys and detonation physics.
Quantum mechanical (QM) algorithms, used on classical computers, can accurately describe the mechanical motions of a compound in its operational environment. But QM scales very poorly with varying molecular sizes, severely limiting the scope of possible simulations. Even a slight increase in molecular size within a simulation can dramatically increase the computational burden. So practitioners often resort to using empirical information, which describes the motion of atoms in terms of classical physics and Newton's Laws, enabling simulations that scale to billions of atoms or millions of chemical compounds.
Traditionally, empirical potentials have had to strike a tradeoff between accuracy and transferability. When the many parameters of the potential are finely tuned for one compound, the accuracy decreases on other compounds.
Instead, the Los Alamos team, with the University of North Carolina at Chapel Hill and University of Florida, has developed a machine learning approach called transfer learning that lets them build empirical potentials by learning from data collected about millions of other compounds. The new approach with the machine learning empirical potential can be applied to new molecules in milliseconds, enabling research into a far greater number of compounds over much longer timescales.
More information: Justin S. Smith et al, Approaching coupled cluster accuracy with a general-purpose neural network potential through transfer learning, Nature Communications (2019). DOI: 10.1038/s41467-019-10827-4
Journal information: Nature Communications
Provided by Los Alamos National Laboratory