Computational method increases design efficiency of protein-based drugs

Researchers from the Institute of Biotechnology and Biomedicine (IBB), in collaboration with scientists from the University of Warsaw recently presented an important update to their AGGRESCAN 3-D computational method, focused on facilitating and reducing the cost of developing new generation protein-based drugs, diminishing their propensity to form aggregates and keeping them stable and active for a longer period of time.
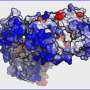
Protein aggregation is a common phenomenon found in a wide range of pathologies, from Parkinson's and Alzheimer's diseases to some cancers and type 2 diabetes. A growing molecular knowledge of this phenomenon has yielded the development of different algorithms capable of identifying and predicting the regions with a greater tendency to aggregate. Among the first was AGGRESCAN, developed by the same researchers of the IBB, which took into account the propensity of the linear sequence, but not the 3-D structure acquired by globular proteins. Four years ago, this same team of researchers expressed the idea of conducting predictions on these protein structures by implementing the AGGRESCAN 3-D (A3D) server. This server offered a higher precision than those based on linear sequencing to predict the aggregation properties of globular proteins. It also provided new features, such as the possibility of easily modelling pathogenic mutations, or a dynamic mode, which allowed modelling the flexibility of small proteins to find potentially hidden regions.
The latest update was presented as a web server freely accessible to the academic world, in addition to a desktop version compatible with Windows, MacOS and Linux. The new algorithm surpasses all previous limitations and substantially broadens computational costs to allow modelling the flexibility of molecules of biomedical interest. It also includes different tools such as an automatic generation of mutations to facilitate redesigns of proteins as antibodies to make them stable and at the same time more soluble, and an improved user interface with which to view the data directly on the website.
"With this update, the A3D becomes one of the most complete aggregation predictors. The fact that one same place offers you the chance to make protein aggregation predictions, model their flexibility, study options for a smart redesign and verify how different factors can affect them, represents a giant step forward with regard to other similar servers," affirms Salvador Ventura, researcher at the IBB and the Department of Biochemistry and Molecular Biology, as well as creator of the A3D. "Among other things, all of this will allow us to improve the production of protein-based drugs, reducing the costs of development, production, storage and distribution."
Protein aggregation, a key element in biomedicine and biotechnology
Protein aggregation has gone from being an ignored area of protein chemistry to becoming a key element within the biomedicine and biotechnology fields. "A bad protein folding and subsequent aggregation is behind a growing number of human disorders and one of the most important impediments to designing and manufacturing proteins for therapeutic applications. These therapies, which imply the use of monoclonal antibodies, growth factors and enzyme substitutions, have already demonstrated high precision of molecular targeting, and therefore the need to study them more in depth becomes even more transcendent," Salvador Ventura concludes.
More information: Aleksander Kuriata et al. Aggrescan3D (A3D) 2.0: prediction and engineering of protein solubility, Nucleic Acids Research (2019). DOI: 10.1093/nar/gkz321
Aleksander Kuriata et al. Aggrescan3D standalone package for structure-based prediction of protein aggregation properties, Bioinformatics (2019). DOI: 10.1093/bioinformatics/btz143
Journal information: Nucleic Acids Research , Bioinformatics
Provided by Autonomous University of Barcelona