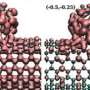
electrodes and a benzene molecule in between: (a) calculated I-V curves; (b) the energy difference calculated by subtracting the DFT energy from the SS-DFT one. The figure shows that SS-DFT predicts the energetically more stable transport state with lower electric currents when compared with the DFT based method. Credit: Zhang Chun")
Figure shows the comparison between SS-DFT and widely used DFT methods for a molecular device consisting of two carbon nanotube (CNT) electrodes and a benzene molecule in between: (a) calculated I-V curves; (b) the energy difference calculated by subtracting the DFT energy from the SS-DFT one. The figure shows that SS-DFT predicts the energetically more stable transport state with lower electric currents when compared with the DFT based method. Credit: Zhang Chun
NUS computational scientists have developed a new version of the density functional theory (DFT) to study nanoscale devices.
Electronic devices are getting smaller and incorporating greater functionality. This is made possible by reducing the size of the electronic components. When their size decreases, the properties of these molecular devices become much more sensitive to their external environment. Computational methods are required to simulate and predict the properties of such small devices. One of them is the density functional theory. These methods are developed from first principles, comprising basic and foundational knowledge which we already know. NUS computational scientists have refined the theory to take into account non-equilibrium effects present during the operation of the devices (e.g. when a battery is connected to a quantum system). This leads to a more accurate and precise prediction.
Prof ZHANG Chun and his Ph.D. student, LIU Shuanglong together with research fellow, Dr Argo NURBAWONO, from the Department of Physics, NUS have developed a more general version of the popular and widely used density functional theory (DFT) that can be applied to steady-state non-equilibrium situations. They introduced an additional degree of freedom, known as the non-equilibrium electron density, into the first-principles modelling. This takes into account the bias-induced non-equilibrium effects when a molecular device operates under a finite bias. This new version of the theory is known as the steady-state DFT (SS-DFT).
The researchers have shown that the widely used DFT in principle is not correct in a steady-state non-equilibrium scenario. In such a situation, two different parameters, the total electron density and the density of current-carrying electrons, are needed to determine the properties of the corresponding non-equilibrium system. The new theory has been implemented into the Spanish Initiative for Electronic Simulations with Thousands of Atoms (SIESTA) computational package to study electronic/ transport properties of various molecular scale devices.
The SS-DFT provides a reliable theoretical tool for understanding and future design of novel molecular-scale devices with enhanced functionality. The SS-DFT-based computational package is now being used by many research groups all over the world. It is used for explaining intriguing transport phenomena observed experimentally at the molecular level and for designing novel types of molecular devices.
The researchers plan to expand the applicability of the theory by including other physical effects, such as electron-phonon interactions and spin-orbital coupling. They also intend to improve the computational efficiency so that it can be used to model large systems around 1,000 atoms.
More information: Shuanglong Liu et al. Density Functional Theory for Steady-State Nonequilibrium Molecular Junctions, Scientific Reports (2015). DOI: 10.1038/srep15386
Journal information: Scientific Reports
Provided by National University of Singapore