How computers are searching for drugs of the future

Drug discovery may bring to mind images of white lab coats and pipettes, but when Henry Lin, PhD, recently set out to find a better opioid with fewer side effects, his first step was to fire up the computers.
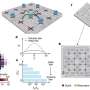
Using a program called DOCK, he uploaded a crystal structure of the opioid receptor found in the brain and accessed a virtual library of 3 million compounds that might bind to a chemical "pocket" on the receptor. Most drugs – from antibiotics to antidepressants – work by binding to specific sites on proteins, but in order to be effective, they must fit just right.
The program spun each compound around, considered the flexibility of its various appendages, and after testing an average of 1.3 million configurations per compound – ranked them by their binding potential. The process, running on computers connected to powerful processors, took about two weeks.
A graduate student at the time, Lin worked with his adviser Brian Shoichet, PhD, professor of pharmaceutical chemistry at the UC San Francisco School of Pharmacy, and Aashish Manglik, PhD, of Stanford University to comb through the top 2,500 compounds for additional factors and selected 23 for experimental testing in living cells – cue lab coats and pipettes.
Increasingly, researchers are turning to virtual experiments for the initial steps of drug development. With ever-faster computers, the early and largely trial-and-error phase of drug development can be reduced to a matter of days, and with ever-expanding online libraries of compounds, drug screens can encompass, literally, all the known chemistry in the world.
Strengths and Limitations
Researchers are cautious about computational drug discovery's potential – only a small fraction of promising compounds actually work when tested in real life – but they say one of its strength is in revealing entirely new compounds as drug candidates.
Shoichet specializes in a popular computational method known as molecular docking. "Where docking fits in is in early discovery research, in finding new departures," he said.
His team's search for the new opioid illustrates both the strengths and limitations of computational drug discovery.
In fact, the initial opioid candidates identified through molecular docking performed only modestly in experimental testing. "Still, the activity they had was highly reproducible and the molecules were highly novel, portending novel biology," said Shoichet.
The team docked another round of compounds with similar structures and tested the top scorers. With collaborators at the University of North Carolina, Chapel Hill and Friedrich Alexander University in Germany, they identified the most potent compound and optimized its pharmacology with computer-guided synthetic elaboration.

That winning compound, named PZM21, is chemically unlike any in current use and may not have been found through more traditional methods. It is a fully computationally-designed compound that is more potent than morphine. In mice, it efficiently blocked pain without the usual side effects of respiratory suppression and constipation and even appeared to be less addictive.
Docking isn't a silver bullet, but it has become a powerful launching point for the long, interdisciplinary process of drug development. Among its major contributions has been the protease inhibitors that have helped make HIV a treatable disease. Researchers are also using docking to screen drug candidates for treating breast cancer, hepatitis C, hypertension, Staphylococcus, the SARS virus and influenza.
Technology Pioneered at UCSF
Molecular docking was pioneered three decades ago by a young UCSF physical chemist named Tack Kuntz, PhD, now professor emeritus at the School of Pharmacy. When Kuntz arrived on campus in the early 1970s, the traditional approach to drug discovery still prevailed.
As Kuntz described it, the process relied on chance and very little theory: "You go out and find new natural compounds and bring them back to test in a lab. Just put chemicals together with an organism and see what happens."
Pharmaceutical chemists hardly gave a thought to the molecular details of how drugs interacted with the body. Many drugs, including the first antibiotics, had been discovered serendipitously, but Kuntz, having seen the new molecular understanding sweeping the field of biology, felt it was time for a similar update in pharmacology.
"The target-based view of biology – that you can understand biology through independent proteins and gene products – had already taken over, but pharmacology was a decade behind," said Shoichet, who was a graduate student in Kuntz's lab in the 1980s.
Kuntz and his colleagues began working toward a more rational approach to drug design in which they tried to identify compounds that could fit specific receptors on proteins, like finding the missing piece of a jigsaw puzzle. In 1982, they published a paper describing the first molecular docking program that could "explore geometrically feasible alignments of ligands and receptors of known structure."
Kuntz sent 10,000 copies of that first docking program to researchers around the country. Soon, other researchers were developing similar computational programs and the excitement quickly spread outside academia. By the 1990s, every major pharmaceutical company had opened a computational drug discovery unit.
Catching Up to an Idea
Despite the initial enthusiasm, however, computational drug discovery didn't lead to rapid results. Kuntz's idea had arrived ahead of its time. It would take decades of incremental advances in molecular biology, imaging and computing technology, before computational drug discovery could begin to fulfill its promise.

A major limitation in the 1990s was the lack of known protein structures. Without these, there were few targets for which to find drugs. In the decades since, thousands of protein structures of possible drug targets have been revealed by X-ray crystallography and nuclear magnetic resonance imaging.
The discovery of the new opioid candidate, for instance, was possible only because of the recently determined crystal structures of G-protein-coupled receptors, a family of proteins that includes the opioid receptor.
Virtual libraries of compounds have also grown exponentially. In 1991, a database might contain 55,000 compounds; now they contain tens of millions. "The scope of the chemistry we're sampling has been going up around the same rate as Moore's Law," Shoichet said. "There's an insatiable hunger for more and more molecules."
Today's docking programs are able to realistically model the atomic-level interactions between a drug and its target, but some tricky details – such as how atomic forces change when a drug molecule displaces water at the binding site – remain ongoing challenges in the field.
Promises and Proofs
Molecular docking isn't the only form of computer-based drug design. At the UCSF Institute for Computational Health Sciences (ICHS), dozens of researchers are exploring myriad computational methods to advance medical research.
Michael Keiser, PhD, a member of ICHS and an assistant professor at the Institute of Neurodegenerative Diseases, is studying drugs that hit many molecular targets at once, as if striking a chord rather than a single note. This multi-target action was long understood to be the cause of unintended side effects, but can also be directed to treat complex diseases.
Only in the early 2000s did researchers come to acknowledge that many existing drugs work through more than one target – antipsychotics, for example, that hit both serotonin and dopamine receptors. They are now intentionally designing drugs to do so.
"For some diseases that don't have treatments yet, maybe it's because there isn't a single protein that you need to turn on or off; what if the drug needs to hit multiple targets instead?" said Keiser, who was a graduate student of Shoichet's.
In his lab, Keiser uses computational methods to identify chemical patterns among drugs that bind to the same set of targets and find new compounds that have matching pharmacology. This computational approach can recognize similarities among compounds that more conventional analyses would miss. Keiser is now looking towards artificial intelligence technology, known as deep learning, for even better pattern recognition.
Even as computational methods take off, their proof is still in the real world—in cells, animal models, and ultimately in the clinic. "For a while it was common to publish papers with predictions about a small molecule's activities, but no actual testing of these predictions, because the experiments to do so were expensive, difficult or esoteric," said Keiser.
As the need for collaboration has become clear, the partnership between computational prediction and wet lab experiments has noticeably strengthened in the last decade, said Keiser. "After all, how can you improve your predictions if you are not sure which are wrong?"
Provided by University of California, San Francisco