October 13, 2016 report
Regioselective hydroarylation of alkynes to make ortho-, para-, and meta- products
 Nature Chemistry (2016). DOI: 10.1038/nchem.2602")
(Phys.org)—One chemically relevant reaction is alkyne hydroarylation, or the addition of an alkyne to an arene to form aryl-substituted arenes. However, most routes for this reaction end up with a mixture of ortho- and para-substituted products.
One solution to this problem is to use a metal catalyst that would coordinate to a substituent on an arene and direct ortho substitution, a process known as chelation-assisted alkyne hydroarylation. This has been done with carboxyl groups. The limitation to this approach is that the subsequent decarboxylation step requires an ortho substituent on the carboxylated arene, thus limiting the range of products available through this method. Additionally, the reaction tends to require harsh conditions further limiting substituent options.
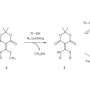
Groups from North Dakota State University and the University of California at Berkeley have devised a catalyst to exact a tandem reaction in which benzoic acid, as their model arene, is used to promote metal-mediated ortho-directing alkyne hydroarylation and subsequent decarboxylation. The carboxyl group is eliminated in situ using the alkene substituent as an activator for metal-mediated carboxyl removal. This reaction worked with various substituents on the benzoic acid, yielding regioselective ortho-, para-, and meta-products. Their work appears in Nature Chemistry.
The first step was to design a catalyst that would promote both the alkyne hydroarylation reaction and the subsequent carboxyl removal. They used benzoic acid and diphenylacetylene as their model reaction and tested several ruthenium(II)-based catalysts as ruthenium(II) is known to promote chelation-assisted carbon-hydrogen bond formation. They also explored other factors such as solvent, temperature, and salt additives to optimize the reaction yield. They eventually landed on Ru(p-cymene)(OAc)2 (10 mol%) as their catalyst, a reaction temperature of 80oC (although this was adjusted for some of the other reactions), and 2:2:1 dioxane/mesitylene/heptane as their solvent.
Zhang, et al. then looked at various commercially available substituted benzoic acids. They report that all of the alkyne hydroarylation reactions using their catalyst and reaction conditions had high stereoselectivity for syn-hydroarylation. They also observed the regioselectivity on the benzene ring that they had hoped would occur if their reaction proceeded with the tandem ortho C-H alkenylation and subsequent decarboxylation.
They observed high yields of the meta-substituted alkenylarenes when conducting their metal-mediated alkyne hydroarylation reaction on a benzoic acid with a strong electron donating groups at the para position. Notably, they observed lower reactivity with electron withdrawing groups in the para position.
Ortho-substituted benzoic acids led almost exclusively to meta-substituted alkenylarenes. Importantly, this supports the fact that their decarboxylation pathway proceeds by coordinating to the alkene rather than because of steric properties.
Zhang, et al. were also able to make regioselective products from meta-substituted benzoic acids. Typically these produce a mixture of ortho- and para-products. In order to obtain exclusively para-products, they had to use sterically hindered meta-substituents.
And, finally they tested several alkynes with 4-methoxybenzoic acid, to determine the versatility of their method. They did not obtain coupled products when testing their catalyst and reaction conditions with terminal alkynes. Unsymmetrical aryl alkynes yielded one product, demonstrating the reaction's regioselectivity for the substitution reaction. Alkynes with simple alkyl substituents required a copper additive to obtain good yields and retrieve the catalyst.
Overall, these results allowed Zhang, et al. to determine that the mechanism for their alkyne hydroarylation reaction was a tandem reaction that involved the metal-coordinated addition of an alkyne group and subsequent decarboxylation of the benzoic acid derivative that was promoted by the alkene substituent. This catalyst opens the possibilities for the synthesis of a variety of aryl alkenes using commercially available benzoic acid derivatives.
More information: Jing Zhang et al. A decarboxylative approach for regioselective hydroarylation of alkynes, Nature Chemistry (2016). DOI: 10.1038/nchem.2602
Abstract
Regioselective activation of aromatic C–H bonds is a long-standing challenge for arene functionalization reactions such as the hydroarylation of alkynes. One possible solution is to employ a removable directing group that activates one of several aromatic C–H bonds. Here we report a new catalytic method for regioselective alkyne hydroarylation with benzoic acid derivatives during which the carboxylate functionality directs the alkyne to the ortho-C–H bond with elimination in situ to form a vinylarene product. The decarboxylation stage of this tandem sequence is envisioned to proceed with the assistance of an ortho-alkenyl moiety, which is formed by the initial alkyne coupling. This ruthenium-catalysed decarboxylative alkyne hydroarylation eliminates the common need for pre-existing ortho-substitution on benzoic acids for substrate activation, proceeds under redox-neutral and relatively mild conditions, and tolerates a broad range of synthetically useful aromatic functionality. Thus, it significantly increases the synthetic utility of benzoic acids as easily accessible aromatic building blocks.
Journal information: Nature Chemistry
© 2016 Phys.org