Scientists simulate the behavior of cell membranes to predict the influence of drugs and toxins on the cell

Scientists from the Moscow Institute of Physics and Technology (MIPT) have developed a highly accurate and relatively fast analytic method that can predict how cell membranes will respond to the molecules of drugs and toxins. This will make it possible to calculate the effect that a medicine will have on cells before any experiments. The research has been published in the Journal of Chemical Theory and Computation.
"The distinctive feature of our method is that it provides us with a complete description of changes in a molecule. We can track positions of all the atoms at once, assigning specific values to every structure variant, which we can use later for statistical analysis," says co-author Ivan Gushchin.
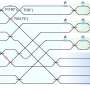
Simulating the behavior of biomolecules is highly problematic, because the movements of every atom have to be described— even a small molecule of 54 atoms requires 156 sets of values. The gigantic amounts of information acquired during the simulation are time-consuming to process and interpret.
The research group led by Gushchin found a method to simplify the analysis of the simulation results without a significant decrease in accuracy. To process the incoming information, scientists used principal component analysis (PCA), a method that selects the most essential data. It reduced the amount of data required for the research by 10 times, with a decrease in accuracy of only 10 percent.

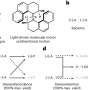
This approach was tested on an experimentally well-studied dioleoylphosphatidylcholine (DOPC) lipid. For the simulation, scientists chose eight different force fields—parameter sets describing the interaction between all the atoms. Some force fields are used for a very detailed description and others for a more approximate one. The researchers then applied the PCA.
They found that the motion description of a 54-atom molecule requires only 14 components—collective movements of a certain atom ensemble. For example, one of the components is responsible for two DOPC molecule hydrophobic tails moving apart in a scissor-like motion.
Lipids are the building material a cell membranes, molecules have to pass through it to affect the cell. This is why studying lipids with single-atom accuracy will help to predict the effect of drugs and toxins on cells and whole organisms in silico, thus accelerating the search for new medicines. Simulation can also be useful in aging research, the mechanism of which is thought to be related to changes in the structure of molecules of cell membranes.
More information: Pavel Buslaev et al. Principal Component Analysis of Lipid Molecule Conformational Changes in Molecular Dynamics Simulations, Journal of Chemical Theory and Computation (2016). DOI: 10.1021/acs.jctc.5b01106
Provided by Moscow Institute of Physics and Technology