Studies reveal a unified approach to combating several bacterial diseases

Researchers at the University of Texas, the University of Connecticut, and the U.S. Department of Energy's (DOE's) Argonne National Laboratory have discovered structural similarities among bacteria of various types that create the possibility of using similar approaches to fight the infections they cause.
The research began by studying Actinomyces bacteria, which commonly cause oral abscesses and gum tissue infections. As it happens, the most odious species of this type must rely on Actinomyces oris (A. oris) bacteria to make attachments to mouth tissues. In this way, the researchers were led to study how A. oris bacteria make their crucial attachments.
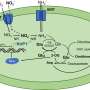
Bacteria secrete a wide variety of proteins, which are large, complex molecules whose ability to recognize and interact with other molecules depends critically on how they fold to become three-dimensional structures with active sites. A. oris bacteria attach to bodily cells and other bacteria by means of two types of protein composites: one type permits the bacteria to adhere to tooth surfaces, and the other is crucial for forming biofilms and bacterial colonies on gum tissues. These stubby hair-like proteins are assembled outside an A. oris bacterium's cell wall using protein pieces secreted in their unfolded, and therefore useless, state. The purpose of the research was to learn enough about how the pieces, called pili proteins, fold upon assembly to formulate antibiotic strategies for disrupting the folding process.
"These pili are very important for virulence, but they must be arranged and folded correctly in order for a pathogen to recognize and interact with a person's bodily cells," said Argonne Distinguished Fellow Andrzej Joachimiak, who collaborated in the research and is the Director of the Midwest Center for Structural Genomics and the Structural Biology Center. "Before we undertook this study, people didn't understand how the proteins folded when being assembled. This is a completely new discovery."
Previous research had shown that pili proteins contain special chemical bonds, called disulfide bridges, that are central to the folding process. The research teams at the University of Texas and the University of Connecticut established that formation of these disulfide bonds is required for pili assembly, cell-to-cell adhesion and biofilm creation by A. oris bacteria. They also showed that disulfide bond formation is likely catalyzed by a versatile enzyme the researchers called MdbA. Further, they demonstrated that MdbA works with another enzyme called VKOR and that the pair possibly comprises a disulfide bond-forming machine that is essential for pili folding and assembly.
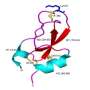
To confirm and extend their findings, the researchers had the MdbA enzyme's structure analyzed at Argonne by means of the protein crystallization technique. The enzyme was crystallized at the Advanced Protein Characterization Facility (APCF). and the resulting crystal was subjected to high-brilliance X-rays at the Structural Biology Center's 19-ID beamline at the Advanced Photon Source (APS). Both the APCF and APS are DOE Office of Science User Facilities located at Argonne.
"We crystallized the A. oris protein and found that it has an active site and a fold of thiol-disulfide oxidoreductase," said Jerzy Osipiuk, a protein crystallographer at the APCF who purified and crystallized the MdbA protein. The significance of this finding is that thiol-disulfide oxidoreductases control the formation of disulfide bonds.
Joachimiak indicated how such a finding could contribute to the development of an effective antibiotic. "The MdbA enzyme is required to maintain bacteria virulence. If we could identify a small ligand that binds to the active site of that enzyme and inactivates it, then the pathogen would not be virulent because pili would be prevented from forming.
"For further development," Joachimiak continued, "we would need to solve the crystal structure of a complex consisting of the MdbA enzyme together with the ligand. Then we could use structure-based methods to redefine the ligand or change its chemical structure. We typically do this collaboratively, by working with laboratories that have large libraries of compounds."
The structural analysis performed at Argonne also showed that the active thiol-disulfide oxidoreductase fold in A. oris's MdbA is a common feature of enzymes that play crucial roles in pili folding in other bacteria. These enzymes in the major human pathogens Staphylococcus aureus, Escherichia coli, and Mycobacterium tuberculosis are very different from the MdbA enzyme in A. oris, but they all contain an active thiol-disulfide oxidoreductase fold. This discovery came about by comparing the MdbA's structure to structures of pili-folding enzymes found in the Protein Data Bank, which archives information about the three-dimensional shapes of proteins, nucleic acids and complex assemblies provided by researchers worldwide. The comparison yielded the insight that infections due to all of these bacteria could perhaps be combated in similar ways.
The researchers next turned to studying pili folding in Corynebacterium diphtheriae (C. diphtheriae) bacteria, which are responsible for often deadly bouts of diphtheria. The pili of this pathogen are responsible for tissue attachments and the toxin the bacteria produce; these pili also start out as unfolded precursors. Structural analysis of the C. diphtheriae's MdbA enzyme showed that it is different from A. oris's MdbA enzyme; nevertheless, both share an active thiol-disulfide oxidoreductase fold and thus may be vulnerable to similar antibiotic treatments.
The APS is the greatest contributor to the Protein Data Bank, having deposited 16,887 solved structures to date, over 5,500 more than the next largest contributor worldwide. That dominance is likely to continue for two reasons. One is that most protein structures were solved without the benefit of the APCF, which is a new facility that houses state-of-the-art robotic laboratories dedicated to the high-throughput production, characterization, and crystallization of proteins and protein complexes. The robotic systems at the APCF can process crystallizations at least 10 times faster than was previously possible, and the facility is linked to the APS, so it can efficiently deliver protein crystals there for X-ray analysis. The second reason is the impending APS Upgrade.
"The APS Upgrade will be incredible," Joachimiak said. "Right now, the X-ray beam at the APS is narrow vertically but wide horizontally, and it needs to be strongly focused to achieve the microbeams we need. After the upgrade, the beam will be small and circular and will produce superbly brilliant micrometer-size beams. Basically, the upgrade will give us the best possible X-ray beam."
More information: Melissa E. Reardon-Robinson et al. A thiol-disulfide oxidoreductase of the Gram-positive pathogen is essential for viability, pilus assembly, toxin production and virulence , Molecular Microbiology (2015). DOI: 10.1111/mmi.13172
Melissa E. Reardon-Robinson et al. A Disulfide Bond-forming Machine Is Linked to the Sortase-mediated Pilus Assembly Pathway in the Gram-positive Bacterium , Journal of Biological Chemistry (2015). DOI: 10.1074/jbc.M115.672253
Journal information: Molecular Microbiology , Journal of Biological Chemistry
Provided by Argonne National Laboratory