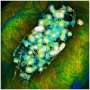
This is the sampling site located at the A.C. and Lela Morris Prairie Reserve, located in Jasper County Iowa. Credit: Jim Tiedje
For soil microbiology, it is the best of times. While no one has undertaken an accurate census, a spoonful of soil holds hundreds of billions of microbial cells, encompassing thousands of species. "It's one of the most diverse microbial habitats on Earth, yet we know surprisingly little about the identities and functions of the microbes inhabiting soil," said Jim Tiedje, Distinguished Professor at the Center for Microbial Ecology at Michigan State University. Tiedje, along with MSU colleagues and collaborators from the U.S. Department of Energy Joint Genome Institute (DOE JGI) and Lawrence Berkeley National Laboratory (Berkeley Lab), have published the largest soil DNA sequencing effort to date in the March 10, 2014, issue of Proceedings of the National Academy of Sciences (PNAS). What has emerged in this first of the studies to come from this project is a simple, elegant solution to sifting through the deluge of information gleaned, as well as a sobering reality check on just how hard a challenge these environments will be.
"The Great Prairie represents the largest expanse of the world's most fertile soils, which makes it important as a reference site and for understanding the biological basis and ecosystem services of its microbial community," said Tiedje. "It sequesters the most carbon of any soil system in the U.S. and produces large amounts of biomass annually, which is key for biofuels, food security, and carbon sequestration. It's an ecosystem that parallels the large ocean gyres in its importance in the world's primary productivity and biogeochemical cycles."
Since the release of the first human genome over a decade ago, the applications of DNA sequencing have been extended as a powerful diagnostic technique for gauging the health of the planet's diverse ecological niches and their responsiveness to change. In this ambitious pilot study launched by the DOE JGI, MSU researchers sought to compare the microbial populations of different soils sampled from Midwestern corn fields, under continuous cultivation for 100 years, with those sourced from pristine expanses of the Great Prairie. The rationale is no less compelling than the original motivations underlying the Human Genome Project.
The Great Prairie soil project is also the kind of demanding initiative ideally suited for the DOE JGI which provided the raw sequencing power to actually do it. Beyond the throughput required to generate enough data, a key factor that makes soil a "Grand Challenge" of biology is that there are precious few reference genomes, "Rosetta Stones," to help sift through these data for the nuggets that may inform important traits like agricultural productivity, carbon cycling, nutrient processing, or disease and drought resistance. Another is the sheer scale of the analyses necessary for the vast amount of raw data. For the Great Prairie soil experiment, the team generated nearly 400 billion letters of code, which amounts to more than 130 human genome equivalents, or 88,000 E. coli genomes.
"This is like shredding the contents of an entire library and reassembling an individual volume out of that massive pile of shreds," said the study's lead author, C. Titus Brown of Michigan Statue University, who uses this analogy for how traditional "shotgun" DNA sequencing of environment samples works. Brown likes to use Charles Dickens' "A Tale of Two Cities," as the particular book in explaining the technique (...it was the age of wisdom, it was the age of foolishness...).
The analytic approach used on the prairie samples was first tried out on a recently characterized data set from the study of the human gut microbiome—the community of microorganisms that live inside us. Brown and his colleague, first author Adina Chuang Howe, deployed a compression method, common with large computer files such as JPEG images conveyed through the internet, that allows a substantial amount of data to be discarded without the actual data content being degraded. Brown calls the technique "digital normalization."

This is one of the Iowa corn field soil sampling sites used to generate data for the Michigan State, DOE Joint Genome Institute, Lawrence Berkeley National Laboratory study published in PNAS. Credit: Jim Tiedje
Having tested it on the gut data set, they applied it to the soil set. "These results still continue to stun me," said Brown. "What this gives us is a 2 to 200-fold decrease in computational requirements for the actual biological analysis."
The key point, Brown said, is that in addition to making hard analysis easier and impossible analysis—soil metagenomes, in particular—approachable, the process dramatically improves genome assembly of difficult organisms, and makes transcriptome assembly (of the RNA molecules that encode proteins expressed by the genome) trivial. Moreover, it offers a data management "democratization" empowering scientists who don't have access to cloud- and high-performance computing, to analyze them.
"I think this can lead to a fundamental shift in thinking," Brown said. "We are actually converting standard, heavyweight approaches in biological sequence analysis to an ultra-efficient streaming approach." Consequently, researchers can devote more resources to extracting science from the noise, as their basic analysis expenditures have dropped.
As for the actual biology of the soil, the analysis is still in the works. But in the meantime, the implications for use of this simple, elegant strategy abound.
Janet Jansson, senior staff scientist from Berkeley Lab's Earth Sciences Division, along with Susannah Tringe, head of the DOE JGI's Metagenome Program, championed the project with the DOE's mission in mind.
"It has been our ambition to improve the ability to link genetic information to ecological function with the potential to yield diagnostic tools for improved soil management, carbon sequestration, ecosystem services, and productivity," said Jansson, who has traveled to the remote expanses of the Arctic to sample the microbial communities coming to life in melting permafrost.
"Metagenomic sequence analysis has provided the means to better understand the function of soil communities in general as well as differences and similarities in composition, diversity, and function in different soil ecosystems."
Digital normalization should enable significant improvement in genome assembly, she said, and provide the critical references to advance future investigations of soils and other complex environments.
"This will help us establish patterns of how genes and organisms evolve in soil, and how these can be used to understand and potentially manage adaptive traits such as greenhouse gas fluxes, carbon stability, and plant disease development," Jansson said. "What we do know now is that soil microbes are responsible for cycling nutrients that are of critical importance for all higher forms of life. The role of soil microbes in carbon cycling is one example that has recently been highlighted due to the importance of microbial-mediated uptake and sequestration of carbon as well as the converse processes of organic matter degradation and release of CO2 and methane to the atmosphere. The relative balance between these processes has enormous implications for the atmospheric carbon budget and subsequent global warming trends."
The bottom line of the PNAS study is that despite 400 billion bases of data, it was still insufficient to interrogate the microbial players in the localized soil sample deeply enough, confirming that much more data are needed to study the content of soil metagenomes comprehensively.
Or, to paraphrase another one of Charles Dickens' characters, said Brown, "please sir, can we have some more...data."
More information: Tackling soil diversity with the assembly of large, complex metagenomes, www.pnas.org/cgi/doi/10.1073/pnas.1402564111
Journal information: Proceedings of the National Academy of Sciences
Provided by DOE/Joint Genome Institute