Sensitive technique for taking RNA inventory of individual cells offers powerful tool
")
Every cell is a hectic messaging center, with thousands of genes churning out messenger RNA (mRNA) transcripts for translation into functional proteins. Accordingly, sequencing the mRNA content of an individual cell can reveal critical insights into that cell's health and physiological state.
Unfortunately, as cells contain only tiny amounts of mRNA, on the order of ten trillionths of a gram, accurate quantitation is a major challenge. By introducing critical improvements to existing techniques, however, a research team led by Hiroki Ueda and Yohei Sasagawa at the RIKEN Center for Developmental Biology has now devised a robust and reproducible approach for surveying the mRNA content of individual cells.
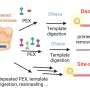
Several whole-transcriptome shotgun sequencing methods are available for performing RNA analyses on large numbers of cells. These high-throughput sequencing technologies, known as RNA-Seq, can be adapted for the single-cell scale by using an enzymatic process called the polymerase chain reaction (PCR) to amplify small amounts of mRNA, but even the most efficient methods tend to be laborious and error-prone. "Multiple PCR assays are required for a single cell, and purification is needed to remove unexpected byproducts," explains Sasagawa, now based at the RIKEN Advanced Center for Computing and Communication. Such inefficiencies lead to inconsistent results, making it difficult to distinguish meaningful differences in gene expression from experimental error.
To overcome these limitations, the researchers designed a dramatically improved single-cell RNA-Seq technique with optimized reaction conditions, and introduced failsafe mechanisms to ensure that mRNA is efficiently amplified without generating confounding artifacts. Their 'Quartz-Seq' method enabled them to characterize the total mRNA content of individual embryonic stem cells (ESCs) with unprecedented reproducibility across experiments.
The Quartz-Seq method could readily distinguish between ESCs and more developmentally mature 'primitive endoderm' cells based on gene expression as with other RNA-Seq methods. However, Quartz-Seq is also able to measure clear differences in gene expression among multiple ESCs at different stages in the cell cycle. The researchers could even confidently distinguish true variations in gene activity among seemingly identical cells, demonstrating the sensitivity of the method. "Global gene expression heterogeneity may contain important biological information about cell fate, culture environment and drug response," says Sasagawa. "We showed that single-cell Quartz-Seq can detect this heterogeneity."
The Quartz-Seq method is already proving valuable for exploring genetic differences among individual cells. In the future, Ueda and Sasagawa hope to further streamline Quartz-Seq and expand their technique to detect other classes of RNA.
More information: 1.Sasagawa, Y., Nikaido, I., Hayashi, T., Danno, H., Uno, K. D., Imai, T., Ueda, H. R. Quartz-Seq: a highly reproducible and sensitive single-cell RNA-Seq reveals non-genetic gene expression heterogeneity. Genome Biology 14, R31 (2013). dx.doi.org/10.1186/gb-2013-14-4-r31
Journal information: Genome Biology
Provided by RIKEN