New molecule heralds hope for muscular dystrophy treatment

(Phys.org) —There's hope for patients with myotonic dystrophy. A new small molecule developed by researchers at the University of Illinois has been shown to break up the protein-RNA clusters that cause the disease in living human cells, an important first step toward developing a pharmaceutical treatment for the as-yet untreatable disease.
Steven C. Zimmerman, the Roger Adams Professor of Chemistry at the U. of I., led the group in developing and demonstrating the compound. The National Institutes of Health supported the work published in the journal ACS Chemical Biology.
Myotonic dystrophy type 1 is the most common form of muscular dystrophy in adults, affecting one in 8,000 people in North America. It causes progressive weakness as the muscles deteriorate over time. There is no treatment available for the disease; though a few measures can help ease some symptoms, nothing can halt their inevitable progression.
"This is a disease that currently doesn't have any treatment, so we have a huge interest in finding therapeutic agents," said graduate student Amin Haghighat Jahromi, the first author of the paper.
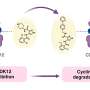
Myotonic dystrophy type 1, called DM1 for short, is caused by a mutation to one gene. In a healthy person, one small segment of the gene – a DNA sequence of CTG – is repeated a few times. In someone with DM1, the sequence is repeated more than 50 times, even up to thousands of repeats. The sequence is transcribed into RNA over and over, like a skipping record stuck in a loop. The repetitive RNA binds to the protein MBNL1, which is essential for regulating protein balance in cells. The RNA traps the MBNL1 protein in aggregates within the cell's nucleus.

"The RNA is functioning in an abnormal way, and unfortunately, it's toxic," Zimmerman said. "MNBL regulates a process called alternative splicing that controls how much of different proteins are made. Affected cells make the proteins, just not at the right levels, so all the levels are imbalanced. There are more than 100 proteins that are affected."
The Illinois group developed a small molecule that could infiltrate the nucleus and bind to the RNA, forcing it to let go of MBNL1 so the protein can do its job. The molecule is small and water-soluble so it can cross the membrane into the cell, which has been a challenge for researchers attempting to use methods with larger molecules. It specifically targets only the repeating RNA sequence so as not to interfere with other cellular functions.
The researchers administered the molecule to live cells that have the disease features of DM1. Using advanced microscopy methods, they were able to watch the cells over time to see how they responded to the molecule. In only a few hours, they saw the clusters within the nucleus break up and were able to measure that the MBNL1 protein had increased its regulatory activity.
"This is the first study that gives direct evidence for the function of the compound," Jahromi said. "We track how the cell is changing upon treatment with the compound and see the effect directly."
Next, the researchers plan to begin collaborating with other groups to test their molecule in fruit flies and mice. Although the molecule will need many rounds of testing for toxicity, efficacy, metabolism and possible side effects before human trials can begin, finding a molecule that works in living cells is an important first step toward making a drug that could treat myotonic dystrophy.
"We're close to developing drug candidates that can be tested in animals. And if it works in animals, then we move hopefully into clinical trials with humans," Zimmerman said. "It's heartbreaking, at one level, to say we're years away from something that's going to be in the clinic. On the other hand, we now have targets. We now know how to go after this disease. It gives patients and their families a bit of hope."
A proof version of the paper, "A Novel CUGexp·MBNL1 Inhibitor With Therapeutic Potential for Myotonic Dystrophy Type 1," is available online.
Journal information: ACS Chemical Biology
Provided by University of Illinois at Urbana-Champaign