December 3, 2012 feature
It's complicated: Hidden protein folding complexity revealed by simple Markov state models
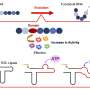
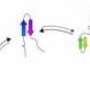
 is shown in black. Full rate matrices with error estimates are given in SI Text. Copyright © PNAS, doi:10.1073/pnas.1201810109")
(Phys.org)—Complex systems often exhibit metastable dynamical behavior – that is, the systems appears to be in an equilibrium state but are actually confined to part of the phase space, while at much longer time scales transition between other such metastable states. (Water provides two well-known examples of metastable dynamical behavior: the delays in both the evaporation of overheated water and the freezing of under-cooled water.) Analysis of this behavior has often focused on stochastic – and more recently, Markov – processes. In particular, Markov State Models (MSMs) have been particularly successful, due largely to their ability to model high-dimensional state spaces, biomolecules, and – again more recently – protein folding kinetics.
Moreover, MSMs have demonstrated quantitative agreement with experimental research. Now, scientists at Stanford University have developed two methods – Flux Robust Perron Cluster Analysis (FPCCA+) and Sliding Constraint Rate Estimation (SCRE) – that allowed them to create accurate rate models from protein folding simulations. The researchers then applied these techniques to a series of massive simulation datasets generated by either Anton (a special-purpose supercomputer for molecular dynamics simulation) or Folding@home, a distributed computing project that runs protein folding, computational drug design, and other molecular dynamics simulations.
, FiP35 (B), and GTT (C) are summarized as networks. Simulated structures are shown in rainbow. For HP35 (A), experimental structures are shown in black (crystal structure 2f4k) and gray (NMR structure 1vii). For FiP35 (B), experimental structures are shown in black (holo PDB: 1i8g) and gray (apo PDB: 1i6c). For GTT (C), the experimental holo structure is shown in black. Full rate matrices with error estimates are given in SI Text. Copyright © PNAS, doi:10.1073/pnas.1201810109")
Professors Vijay S. Pande and Yu-Shan Lin, lead researcher Kyle A. Beauchamp, and researcher Robert McGibbon faxed a range of challenges in designing and executing their study. "The main challenge in developing new analysis methods like Flux Robust Perron Cluster Analysis and Sliding Constraint Rate Estimation was to understand the limitations of the current generation of tools," Pande tells Phys.org. "Once we identified the limitations of our current analysis methods, we could then develop ways to improve upon them," Pande continues. "The challenges of working with large datasets are something that we always keep in mind."
In fact, he adds, the team has developed their MSMBuilder software with such datasets in mind – so applying their methods to simulations from Folding@Home or Anton is generally straightforward. "Moving forward, we hope to develop future methodological advances that further increase our ability to model protein folding. I think we really focus on a two-step process that involves a lot of thinking about both methods and their application to biophysics."
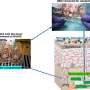
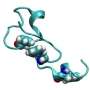
. Near-native (gray: 361 K, rainbow: 300 K) conformations are compared for both HP35 datasets. The crystal structure (PDB:2f4k) is shown in black. (B). A 2D RMSD histogram suggests that the 300 K simulations populate both the native and near-native states, with a population ratio of approximately 9∶1. The line x ¼ y was used to separate the N and N’ states. Copyright © PNAS, doi:10.1073/pnas.1201810109")
Expanding on how their findings suggest that some beta containing proteins can form long-lived native-like states with small register shifts, Pande notes that the presence of register shifts in multiple systems was a surprising and interesting discovery. "While we knew that such register shifts were possible, it was quite interesting to see them pop up across the whole spectrum of beta proteins in the dataset."
Regarding their view that their results demonstrate that even the simplest protein systems show folding and functional dynamics involving three or more states, Pande points out that their models help reconcile the apparent gap in complexity between protein folding simulation and experiment. "For the systems we studied, we detected multiple states," he notes. "However, we also found that the many-state behavior sometimes involves only small populations."
The scientists also see other areas of research that might benefit from their findings. "In the future," Pande concludes, "we hope similar methods could be used to understand protein dynamics in human disease."
More information: Simple few-state models reveal hidden complexity in protein folding, PNAS, October 30, 2012 vol. 109 no. 44 17807-17813, doi:10.1073/pnas.1201810109
Journal information: Proceedings of the National Academy of Sciences
Copyright 2012 Phys.org
All rights reserved. This material may not be published, broadcast, rewritten or redistributed in whole or part without the express written permission of PhysOrg.com.