August 30, 2011 feature
Water, water everywhere: Polarization dramatically affects H2O structure revealed through molecular dynamics simulation
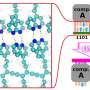
 that shows the density at each volume element centered at every point in space defined by coordinates (x, y, z). It suffers from the severe difficulties associated with viewing and analysis of a general three-dimensional map. Most commonly used is the one-dimensional radial distribution function (RDF) that averages the number of O or H atoms over all volume elements at a distance r from the center of C60, where r2 Ľ x2 þ y2 þ z2. Although such averaging increases the signal-to-noise ratio, the RDF hides many features of the three-dimensional density map. Faced with these limitations, we introduce the azimuthal distribution function (ADF) that calculates density maps in thin spherical shells at a particular r value in terms of spherical polar coordinates (r, θ, φ). The results, which present all salient three-dimensional features of C60, are easily viewed on paper using the Sanson-Flamsteed projection (69) used by early cartographers. © PNAS, doi: 10.1073/pnas.1110626108")
(PhysOrg.com) -- Water is essential to more than its myriad roles in biological, chemical, geological, and other physical processes. Having a precise description of water’s structure is critical to constructing accurate simulations of molecular events, including protein folding, substrate binding, macromolecular recognition, and complex formation. An important step forward in creating such a description has been demonstrated at Stanford University School of Medicine, where researchers discovered that polarization increases ordered water structure. Their findings will have a significant impact on biological processes.
Dr. Gaurav Chopra and Professor Michael Levitt in the Department of Structural Biology used molecular dynamics simulations involving a state-of-the-art Quantum Mechanical Polarizable Force Field (QMPFF3) to study the hydration of buckminsterfullerene, the smallest hydrophobic nanosphere widely referred to as a buckyball or C60. (Hydrophobic molecules like fullerenes are repelled by water, and tend to be both nonpolar and electrically neutral.)
There were many challenges to overcome in designing and implementing QMPFF3-based molecular dynamics simulations – especially to study the behavior of water molecules next to hydrophobic surfaces at atomic detail and subpicosecond time resolution. “The first was the need to use a suitable polarizable force field,” explains Chopra. “Several exist – for example, AMOEBA, polarizable versions of OPLS, AMBER, and CHARMM – but all of these are empirical having being parameterized to fit experimental data as first laid out by Warshel and Lifson in a 1968 paper discussing their consistent force field. We wanted to use an ab initio force field that would be less sensitive to arbitrary parameterization.” While such a force field had been developed by Algodign, LLC in Moscow, it was not available academically. However, by visiting Algodign in Russia three years ago, and with Levitt’s intervention, they obtained academic rights.
“We started out by adapting the QMPFF3 program, AlgoMD, to work on the multiple cores on our Linux supercomputer (BioX2)” Chopra continues. “Then various tests needed to be done to get the correct equilibration protocol set-up with the correct set of parameters for normal temperature and pressure control along with the most relevant atom type selection for the buckyball to use. Choosing the optimum test system was easy as the Levitt lab had previously worked on this molecule with non-polarizable empirical force fields.”
The final challenge was to find a method to visualize water structure around buckyball. “The most popular method to study the water structure around any solute used a one-dimensional radial distribution function that averages the number of water O and H atoms at a particular distance to increase the signal to noise ratio,” Chopra explains. “This one-dimensional radial distribution function hides many features of the three-dimensional density map around any arbitrary shaped solute. Faced with these limitations and in order to account for the soccer ball symmetry of C60 we introduced the Azimuthal Distribution Function to visualize the O and H density distributed as it is in concentric spherical shells.”
To meet these challenges, the team depended on a long history of related research. “Since the pioneering work on the consistent force field developed by Shneior Lifson more than 50 years ago, the model of an atom has been a nucleus with a partial charge. We believe the time has come to move to a more realistic representation of an atom as a nuclei and an exponentially distributed zero mass electron cloud around it.” The implementation of this representation in QMPFF3 allowed the effect of polarization to be correctly modeled. “We studied the structure of polarized water around polarized Buckminsterfullerene to show that polarization induces a strong hydrophobic effect; this has been under-represented by the limitations due to approximate modeling of atomic interactions in the empirical force fields widely used for the past decades.”
The sensitivity of their novel method for detecting surface roughness shows that the hydrophobic effect is much stronger at short- and long-range for QMPFF3 compared to empirical force fields simulations. For this reason, QMPFF3 is expected to have a profound effect in understanding key biological processes like protein folding. “Using a novel and highly sensitive method to measure surface roughness and detect water ordering, we show that accurate modeling of solute and solvent polarization results in a stronger hydrophobic effect,” Chopra summarizes.
. SDFs for water oxygen (O) and hydrogen (H) density around C60 for (A and B) QMPFF3 and (C and D) OPLS-AA with SPC water (OPLSAA-SPC). The orange contours represent higher O and H atom density, and black contours represent lower density than bulk. Both OPLSAA-SPC and QMPFF3 have an excluded volume around C60, a layer of low water density, and well-defined first and second water hydration shells. QMPFF3 water shells around C60 are more structured in radial and azimuthal direction with well-defined peaks; there is no structure apparent in the azimuthal direction for OPLSAA-SPC. The ratio of highest O to H density was 1.4 for OPLS-AA with SPC water and 1.8 for QMPFF3. © PNAS, doi: 10.1073/pnas.1110626108")
Apart from the azimuthal distribution functions developed to analyze the results, another challenge was to make suitable choices in the simulation protocol to significantly enhance the physical reality of the C60–water system. “The van der Waals equivalent for QMPFF3 is the combination of exchange and dispersion terms in QMPFF3. We used aromatic atom types for C60 that were reparameterized by simple model correction using coupled-cluster with single and double and perturbative triple excitations data in QMPFF3. We also used long-range dispersion correction terms for total energy and pressure caused by truncation of dispersion forces.” Chopra stresses that the study was computationally very intensive, and would not have been possible without the National Science Foundation-supplied BioX2 supercomputer.
Chopra also points out that while QMPFF3 is one of the best polarizable force fields available today, as it is a general purpose ab initio force field which has been parameterized using only quantum mechanical data to successfully reproduce the experimental data for a large array of chemical compounds in all three phases of matter, it is not perfect. “We can reparameterize certain special atom types using a higher level and more accurate quantum mechanical data as well as introduce new atom types for specific applications. The functional form may also need to be modified to further increase the physically realistic representation of the non-bonded parts of the force field currently modeled as dispersion, exchange, electrostatics and induction,” he notes. “These advances could significantly improve the performance of this state-of-the-art polarizable force field.”
On the other hand, Chopra points out, “By adapting the QMPFF3 program on GPUs one could significantly increase its computational performance to study much larger systems of interest at biologically relevant timescales. Based on our tests, QMPFF3 is about 10 times slower than the empirical force field simulations to study protein-water systems on commodity clusters. We therefore think it is important to make advances to simultaneously improve the physical reality as well as increase the computational efficiency of the current state-of-the-art polarizable force field.”
Chopra sees the team’s findings as relevant to a wide range of possible applications. “Our work is at the intersection of material science, nanotechnology and fundamental interactions in protein folding. The nature of the hydrophobic effect forms the basis of protein folding simulations and fullerenes are perfect model systems to study the affect of such interactions. Moreover, polarization has always been neglected or modeled incorrectly – but our results show the importance of polarization resulting in stronger short- and long-range hydrophobic interactions.”
Chopra acknowledges that while their findings are not directly applicable to the development of fullerene-based biosensors as such, biosensors are made using water-soluble fullerene derivatives. “Having discovered the correct way to include of polarization for your system of interest to make it physically realistic could significantly advance the selection of suitable groups to be attached to fullerenes for many applications, including biosensors, as well as for significantly advancing the process of drug discovery. Our result can be used as a quick way to include the effect of the arrangement of water molecules based on the surface topology of a hydrophobic binding pocket. In general, the accurate treatment of polarization to include the affect of solvent in the binding pocket will potentially be useful for advancing computational drug design.” Chopra is also very interested to study the effect of polarization on biological systems like proteins in non-homogenous solvent simulations.
“Ours is a very general technique and any system can be studied with the simulation and analysis methods of this paper, Chopra concludes. “Since QMPFF3 is a general-purpose polarizable force field and, for studying any system, it gives a physically realistic treatment to include polarization, which is essential for any biological system as they are always present in a polar medium like water. Also, our method to study water structure is a significant advance over currently used techniques and should be used to visualize water structure around any arbitrary shaped solute.”
More information: Remarkable patterns of surface water ordering around polarized buckminsterfullerene, Published online before print August 15, 2011, doi:10.1073/pnas.1110626108 , PNAS August 15, 2011
Copyright 2011 PhysOrg.com.
All rights reserved. This material may not be published, broadcast, rewritten or redistributed in whole or part without the express written permission of PhysOrg.com.