Human-cell-derived model of ALS provides a new way to study the majority of cases
For decades, scientists have studied a laboratory mouse model that develops signs of the paralyzing disease amyotrophic lateral sclerosis (ALS) as they age. In a new study appearing in Nature Biotechnology, investigators at Nationwide Children's Hospital have developed a new model of ALS, one that mimics sporadic ALS, which represents about 90 percent of all cases.
ALS, commonly known as Lou Gehrig's disease, is characterized by the death of motor neurons, which are muscle-controlling nerve cells in the spinal cord. As these neurons die, the body's voluntary muscles weaken and waste away; death within five years of diagnosis is common. Only about 10 percent of ALS cases are familial meaning the disease runs in the family. The majority of ALS cases are sporadic, with no family history.
Mutations in the SOD1 gene are found in about one-fifth of people with familial ALS, and for decades, experts have theorized that the gene holds clues to sporadic ALS. Laboratory mice carrying human SOD1 mutations develop signs of ALS as they age, and have been widely used to investigate the causes and potential treatments for the disease. At the same time, however, researchers have questioned whether SOD1 mice are useful and whether SOD1 itself is relevant for understanding sporadic ALS. While dozens of potential therapies have shown promise in the mice, most have failed in patients.
"The mouse models capture a type of familial ALS that accounts for only two percent of all cases. The field has begged for new disease models that can provide a clear window into sporadic ALS," said senior author Brian Kaspar, PhD, principal investigator in the Center for Gene Therapy of The Research Institute at Nationwide Children's Hospital.
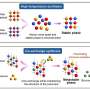
Nationwide Children's researchers attempted to develop such a model by isolating cells from patients' spinal tissue within a few days after death.
First, the team isolated neural progenitor cells from post-mortem spinal tissue of patients with ALS. Neural progenitor cells are the "parent" cells of neurons and astrocytes, cells of the central nervous system. They then coaxed these progenitor cells to develop into astrocytes. Next, the team combined the patient-derived astrocytes with mouse motor neurons. At first, the motor neurons grew normally, but after four days, they began to degenerate. By five days, the number of motor neurons reduced by about half compared to motor neurons that had been grown with control astrocytes. Similar results were seen when the motor neurons were grown with astrocytes from a patient with familial ALS, or with a cell culture broth that had been conditioned by astrocytes from any of the ALS patients. This suggests the ALS-derived astrocytes are releasing one or more unknown toxins.
Further experiments revealed that inflammatory responses and SOD1 may play a critical role in this toxicity. These results suggest that replacing astrocytes may be just as important as replacing motor neuron lost to the disease and that astrocytes and SOD1 need further investigation as targets for therapy.
"It has been a long road, but the hard work starts now," said Dr. Kaspar. "We still need to confront fundamental questions about what is happening to astrocytes and how they are killing motor neurons. And the ultimate goal is to identify therapies that will translate into helping humans."
Provided by Nationwide Children's Hospital