Catalyst-where-you-want-it method expands the possibilities for new drug development
Chemists at The Scripps Research Institute (TSRI) and the Shanghai Institute of Organic Chemistry have described a method for creating and modifying organic compounds that overcomes a major limitation of previous methods. The advance opens up a large number of novel chemical structures for synthesis and evaluation, for example, as candidate pharmaceuticals.
The new method was designed to avoid an unwanted side effect—a diversion of a catalyst molecule to the wrong location—that prevents chemists from manipulating many organic compounds in the class known as heterocycles, which are commonly used as drugs.
The newly described technique gets around this obstacle by generating a reactive catalyst at precisely the desired site on a molecule to be modified.
"We have already applied this technology to enable the modification of a wide range of chemical structures, including a complex drug candidate being developed by a major pharmaceutical company," said Jin-Quan Yu, professor of chemistry at TSRI.
Yu and his colleagues describe the new method in a paper published by the journal Nature on November 10, 2014.
Small Changes with Big Consequences
Heterocycles have the basic carbon-ring structures of other organic compounds, but with one or more of the carbon atoms replaced by a different atom, such as nitrogen or sulfur. Such a seemingly slight change can radically alter the properties of a compound—conferring greater solubility, for example, which is why heterocyclic structures are often preferred in modern synthetic drugs and also are found frequently in natural compounds that are used as drugs.
But heterocycles pose a particular challenge for those who would modify them. In the pharmaceutical industry, for example, chemists typically select a compound that has a desired chemical activity and then attempt to "tweak" it in various ways to optimize its properties and perhaps create useful new properties. The fastest way to do this is by removing a hydrogen atom from one of the compound's backbone carbon atoms and replacing it with a new functional group.
Chemists in recent years have devised various techniques to make this basic type of modification, known as C-H functionalization or C-H activation. Yu's laboratory has been responsible for some of the more powerful ones. But C-H activation techniques generally rely on the use of a metal-containing catalyst molecule to cleave a carbon-hydrogen bond to make way for the new functional group. In heterocycles, a "hetero" (non-carbon) atom such as nitrogen is apt to draw the catalyst away from the targeted C-H bond, thus preventing the desired modification.
"This detrimental effect seriously limits the diversity of drug candidates that can be made via C-H activation reactions," said Yu.
The Right Place at the Right Time
In the new study, Yu's group, working with collaborators in the laboratory of Hui-Xiong Dai (a former member of the Yu group at TSRI) at the Shanghai Institute of Organic Chemistry, found a way around this problem.
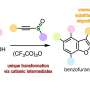
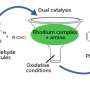
The key to their solution is a molecule that Yu and his TSRI colleagues first described in a 2008 paper, a "directing group" derived from carboxylic acid and known as an N-methoxy amide.
Normally, a directing group facilitates a C-H activation by helping a new functional group into the correct position. In this case, the directing group also reacts with a supplied palladium-containing molecule and oxygen from the air to make the desired palladium catalyst.
"In other words, the directing group generates the catalyst just where it should be on the structure, and at a safe distance from any heterocyclic atom, such as nitrogen, to which the catalyst otherwise would bind and get poisoned or activate C-H bonds at the undesired positions," said Yu.
The team demonstrated the effectiveness of the technique by using it to modify a wide variety of heterocyclic structures that are frequently present in drug molecules, including furans, benzofurans and benzothiophenes; indole, pyrrole, thiazole, pyrazole and imidazole; pyridines; quinoline, pyrazine, pyrimidine, pyrazole and thiazole. The team also used the technique as part of a simple, versatile process for making lactams, a class of heterocyclic compounds that include penicillins.
Yu and his laboratory and collaborators now hope to expand the utility of the new method and explore specific applications in drug discovery.
More information: Overcoming the limitations of directed C–H functionalizations of heterocycles, DOI: 10.1038/nature13885
Journal information: Nature
Provided by The Scripps Research Institute