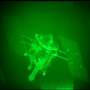
Folding@home simulates activation of key cancer protein, could lead to novel drug design

Once again, computer screensavers have flexed their muscles and solved a great mystery of biology.
For the first time, Folding@home, the Stanford-run program that taps the idle processing time of 200,000 personal computers to elucidate the three-dimensional shape of proteins, has simulated a protein transitioning between its inactive and active configurations.
The particular protein simulated by the research group, Src kinase, plays an important role in many cancers, and this work provides a novel opportunity for designing future drugs to combat its role in disease.
The study, which was led by senior authors Vijay Pande, founder of Folding@home and a professor of chemistry, structural biology and computer science at Stanford, and Benoît Roux, a professor of biochemistry and molecular biophysics at the University of Chicago, was recently published in the journal Nature Communications.
The general role of kinases is to act as an intracellular "molecular switch" that activates other proteins, enabling the cell to carry out its normal duties. Kinases play a particularly important role in regulating cellular growth. Cancer cells corrupt this process and rev up kinase production and activation, causing the cancer to grow and spread unchecked.
To date, there have been relatively few cancer drugs that have successfully targeted and inhibited kinases. The trick is to hit the disrupted kinase at the root of the cancer and to turn it off, without affecting many other similar kinases that are critical, for instance, to heart or kidney cells.
The Src kinase has a specific three-dimensional structure and, like many proteins, its entire structure reconfigures as the protein transforms between its inactive and active states. Scientists can determine the exact structure of these two states using a technique called x-ray crystallography.
These images provide the blueprints for researchers to design drugs that interfere with the protein when it is running out of control. Unfortunately, these two states are fairly similar across the kinase family, making it difficult to target only the kinases being exploited by the cancer.
On the other hand, while the various configurations that Src kinase takes during its conformational transition between active and inactive could prove useful, those short-lived states are nearly impossible to detect experimentally. But unlike x-ray crystallography, the simulations are able to provide a view of these intermediate conformations. These could prove useful.
"The intermediate states could be differentiable from other kinases," said Pande, a co-principal investigator at the Simbios Center for Biomedical Computation at Stanford. "We haven't proven it yet, but these intermediate targets might present the opportunity to design more selective drugs."
The program predicts these states by combining elegant algorithms with the brute force of the graphics processing units of 200,000 computers, providing more than 33 petaflops of processing power.
The algorithm knows only the protein's start and end configurations – the active and inactive states – and discovers the various ways the protein could rearrange itself to get from one end-state to the other. As it runs, certain transitions occur more frequently, increasing the likelihood that they are on the actual path the protein follows in real life. Once the simulation has run enough times, the scientists can identify, with statistical certainty, the most likely order and shape of the transition.
The next step, Pande said, is to conduct experiments that will confirm the existence of these intermediate states.
"If this is really correct, it's a piece of the puzzle that nobody had before," Roux said. "This is one of the first times that computation can give you something that you can almost not get from pure experiment. It would certainly shake things up from a drug development standpoint."
Pande thinks that this work indicates that Folding@home has become robust enough to move beyond just identifying protein structures, toward simulating how all sorts of molecular interactions occur. A strength of the program, he said, is the length of the calculations it can carry out.
Typically, a supercomputer can crunch out 100 nanoseconds, or maybe a microsecond, of continuous data simulations. By splitting the same work over hundreds of thousands of computers, though, Folding@home can compute a few hundred microseconds, as was the case with Src Kinase.
"If you row a boat from Europe to America, and you can only go a mile, you're never going to discover anything," Pande said. "But if you can get a few thousand miles off shore, you'll see something exciting and different from what you've seen before."
More information: "Activation pathway of Src kinase reveals intermediate states as targets for drug design." Diwakar Shukla, Yilin Meng, Benoît Roux, Vijay S. Pande. Nature Communications 5, Article number: 3397 DOI: 10.1038/ncomms4397 . Received 22 July 2013 Accepted 06 February 2014 Published 03 March 2014
Journal information: Nature Communications
Provided by Stanford University