Scaling Goes eXtreme: Researchers reach 34K CPUs

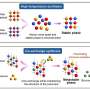
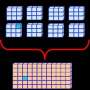
(PhysOrg.com) -- Currently, researchers have demonstrated the scalability of high-level excited-state coupled-cluster approaches and parallel-in-time algorithms, reaching a staggering 34,000 Core Processing Units. Researchers at Pacific Northwest National Laboratory are targeting the software that is capable of describing the behavior of molecules in excited states, as well simulating their dynamics.
To effectively attack major scientific problems in energy, the environment, health, and national security, state-of-the-art algorithms need to be developed that are capable of running effectively on emerging computer systems capable of performing more than one quadrillion operations per second (known as a petaflop). The development of advanced algorithms that will perform on these petascale—and on soon-to-be exascale—computer architectures will make it feasible to perform massive modeling and simulation calculations on these types of computers.
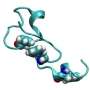
PNNL researchers also are looking toward integrated multiscale approaches that can be used to model chemical processes in realistic settings and that can capitalize on having highly scalable implementations of electronic structure methodologies. This will allow high-level description of large molecular systems in realistic settings defined by finite temperatures and pressures. "We think this method will significantly enhance our understanding of fundamental processes in solar cells, systems mimicking photosynthesis, and optically active materials," says PNNL researcher Karol Kowalski.
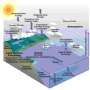
Progress in this field will significantly enhance the systems-size limit that can be managed by computer simulations and will set new standards for accuracies attainable in molecular simulations. In particular, researchers will use new, highly scalable codes to describe the energy conversion in light harvesting (photosynthesis) molecular systems and to simulate the structure, dynamics, and reactions at the mineral (Fe2O3)/ solution interface as a function of pressure and temperature.
Significant progress in scalability of a class of methodologies known as non-iterative CC methodologies for excited states was accomplished thanks to several factors, including redefined local memory management, a new global addressing strategy used to handle large data sets stored on Global Arrays, and more efficient ways of dealing with complex large-data expressions defining the CC equations. These improvements significantly increased the performance of the active-space version of non-iterative CC methods accounting for important, from the point of view of obtained accuracies, triply excited configurations.
Researchers continue to develop next-generation algorithms for massively parallel computers. Researchers are planning to address the following problems:
• Find an efficient solution to local memory bottlenecks characterizing correlation effects at different orders, further optimize the communication pattern in coupled-cluster approaches for excited states, and characterize the performance of resulting codes on computers with 50K-100K of CPUs.
• Development of the exascale parallel in time algorithms for use with terascale ab initio molecular dynamics and the terascale molecular dynamics programs
• Development of efficient interface between ab-initio theories and an adaptive multiscale simulation module.
"At the completion of this project, we expect to have a suite of massively parallel tools to perform excited-state calculations for molecular systems composed of hundreds of atoms and new algorithms to perform dynamic simulations for much longer propagation times," says Kowalski. "Another important outcome is closely related to the development of multiscale approaches capable of incorporating the changes in chemical structure of the surrounding environment."
More information: References:
-- Kowalski, K., Krishnamoorthy, S., Villa, O., Hammond, J., N. Govind, 2010. Active-space completely-renormalized equation-of-motion coupled-cluster formalism: Excited-state studies of green fluorescent protein, free-base porphyrin, and oligoporphyrin dimer, The Journal of Chemical Physics, April 16, 2010, doi:10.1063/1.3385315
-- E.J. Bylaska et al. Large Scale Plane Wave Density Functional Theory: Formalism, Parallelization, and Applications" in Computational Methods for Large Systems: Electronic Structure Approaches for Biotechnology and Nanotechnology, (John Wiley & Sons, Inc.) Ed. J.R. Reimers. (in press).
-- K. Kowalski, M. Valiev, Journal of Chemical Physics 131, 234107 (2009).
Provided by Pacific Northwest National Laboratory