Using fish to illuminate the architecture of inherited disease
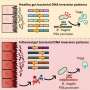
A research team led by scientists from the Duke University Medical Center has developed a way to simultaneously look at the effects of 125 mutations occurring on 14 different genes. They used zebrafish as a model to analyze the function of every known mutation in an inherited syndrome called BBS, Bardet-Biedl Syndrome.
Being able to analyze the functions and interactions of all mutations in a complex inherited disease could have implications for a broad range of disorders. The study found that, while mutations at one of at least 14 genes are responsible for the disorder, mutations elsewhere in the genome may modify the severity and diversity of the symptoms.
"The human genome project and new technologies can help us identify mutations in patients' genomes, but the challenge is how to interpret such variation and how to use it to improve the ability to predict what this means with respect to a patient's clinical presentation," said senior author Nicholas Katsanis, Ph.D., Jean and George Brumley Jr., M.D., Professor of Developmental Biology, Professor of Pediatrics and Cell Biology, and Director of the Duke Center for Human Disease Modeling. "Our work demonstrates that it is possible to develop functional bioassays using a vertebrate model that predicts whether a mutation has a role in a complex disease, like Bardet-Biedl syndrome, which we studied."
The study was published online during the week of May 24 in the Proceedings of the National Academy of Sciences.
BBS is an interesting disease to use as a study model because it involves a number of different traits that are highly variable among patients, said Katsanis, whose endowed professorship is in the Neonatal-Perinatal Research Institute at Duke. People with BBS may have retinopathy, obesity, mental retardation, more than the usual number of fingers or toes, and other distinct traits. BBS has become something of a workhorse for understanding variability of disease in humans, he said.
Simultaneously studying all the mutations in BBS led to some notable discoveries. Contrary to popular scientific belief, some mutations in BBS not only cause the loss of function of a protein, they actually influence the "good" remaining copies of the protein. In addition, the researchers saw that a subset of commonly occurring versions of some genes (called alleles) can be detrimental to protein function. The common alleles also can interact with strong, rare alleles to determine a trait.
"We speculate that such interactions are probably widespread across genetic disorders," Katsanis said. "Indeed, this might help settle a 100-year-old argument about common versus rare mutations and how they might underlie human genetic disorders. Perhaps not surprisingly, the answer is both, in a context-dependent fashion."
Katsanis is a world expert in ciliopathies such as BBS, in which the primary cilium (protrusion) of cells is abnormal and leads to a host of problems. About one child in 1,000 live births will have a ciliopathy, an incidence that is in the range of Down's syndrome, said Katsanis.
Katsanis said that the complex architecture of BBS probably is not unique to this disorder so the approach used by these researchers could improve understanding of a wide variety of human traits.
The researchers did in vivo tests in fish to learn whether they would develop defects if they had specific mutations and then validated their results with in vitro tests on cells in a lab dish to learn whether the aberrant activity in zebrafish embryos could be explained by defective behavior in mammalian cells.
Importantly, by comparing their data with previous clinical studies, they found their tools to be both highly sensitive and highly accurate, correctly predicting the effect of mutations at 98 percent, with a false-positive rate of less than 10 percent. "These numbers are quite critical, because they mean that we can use this approach to interpret information in the clinical setting; these percentages should be good enough for application in clinical labs," Katsanis said.
"A next step is to develop similar tools to let us evaluate various human genetic mutations within the context of their functions," Katsanis said. "Genotype must have a predictive value or it doesn't tell us much. Knowing all of the disease-related variants in a genome is only a starting point, because our work suggests that there is complexity that many do not yet appreciate in disease architecture."