Delaying the aging process protects against Alzheimer's disease

Aging is the single greatest risk factor for Alzheimer's disease. In their latest study, researchers at the Salk Institute for Biological Studies found that simply slowing the aging process in mice prone to develop Alzheimer's disease prevented their brains from turning into a neuronal wasteland.
"Our study opens up a whole new avenue of looking at the disease," says the study's leader, Howard Hughes Medical Investigator Andrew Dillin, Ph.D., a professor in the Salk Molecular and Cell Biology Laboratory. "Going forward, looking at the way we age may actually have more impact on the treatment and prevention of Alzheimer's disease than studying the basic biology of the disease itself."
Their finding, published in the Dec. 11, 2009 issue of the journal Cell, is the latest clue in the Salk scientists' ongoing quest to shed light on the question of whether Alzheimer's disease onset late in life is a disastrous consequence of the aging process itself or whether the beta amyloid aggregates that cause the disease simply take a long time to form.
Age is the major risk factor for the development of Alzheimer's disease. Beyond age 65, the number of people with the disease doubles every five years. Centenarians, however, seem to escape most common age-related diseases, including the ravages of Alzheimer's disease.
"In this study, we went directly to the root cause of Alzheimer's disease and asked whether we could influence the onset of the disease by modulating the aging process," says first author Ehud Cohen, Ph.D., formerly a postdoctoral researcher in Dillin's lab and now an assistant professor at the Hebrew University-Hadassah Medical School in Jerusalem, Israel.
To answer this intriguing question, he slowed the aging process in a mouse model for Alzheimer's by lowering the activity of the IGF-1 signaling pathway. "This highly conserved pathway plays a crucial role in the regulation of lifespan and youthfulness across many species, including worms, flies, and mice and is linked to extreme longevity in humans," he explains. As a result, mice with reduced IGF-1 signaling live up to 35 percent longer than normal mice.
Cohen then employed a battery of behavioral tests to find out whether it was simply the passage of time or aging per se that determined the onset of the disease. Chronologically old but biologically young animals appeared nearly normal long after age-matched, normal-aging Alzheimer's mice exhibited severe impairments in their ability to find a submerged platform in the Morris water maze or stay atop a revolving Rota Rod.
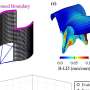
"These behavioral differences between normal and long-lived mice were apparent at nine months of age, but the big surprise came when we took a closer look at the plaques in their brains," says Cohen.
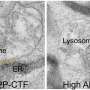
One of the telltale signs of Alzheimer's disease is the buildup of toxic clumps of beta amyloid plaques in the brain. Beta amyloid production probably occurs in all brains, but healthy cells clear away excess amounts. Brains of people with Alzheimer's disease, on the other hand, are unable to control beta amyloid accumulation. The same is true for Alzheimer's mouse models, which are genetically engineered to overproduce beta amyloid.
Although long-lived mice didn't show any of the cognitive or behavioral impairments typical of Alzheimer's disease till very late in life, their brains were riddled with highly compacted plaques.
"Although before it was thought that plaques are the causative agents of Alzheimer's disease, our results clearly support the emerging theme that they have a protective function," says Cohen. "As mice age, they become less efficient at stowing away toxic beta amyloid fibrils in tightly packed aggregates."
An earlier study by Cohen, Dillin, and colleagues, in which they had used roundworms to study the effects of the aging process on protein aggregation, had indicated that high molecular weight aggregates of beta amyloid might actually be less toxic than smaller beta amyloid fibrils. "But worms don't have brains as we do, and it wasn't clear whether these results would be relevant for mammals," he says.
And what about those lucid centenarians? "Interestingly, three studies found that some very long-lived humans carry mutations in components of the IGF-1 signal pathway—the same pathway that we perturbed to increase the lifespan of the mice in our study," says Dillin.
"The reporting of this work is a celebration for the entire field of aging researchers, as it validates the long-held hypothesis that genetic and pharmacologic changes to create a healthy lifespan, or 'healthspan,' can greatly reduce the onset of some of the most devastating diseases that afflict mankind," he adds.
Source: Salk Institute (news : web)